天然有机质和金属离子在矿物表面的共吸附
2018-09-03严玉鹏王小明冯雄汉谭文峰
易 层,严玉鹏,王小明,胡 震,熊 娟,刘 凡,冯雄汉,谭文峰
(华中农业大学资源与环境学院,农业部长江中下游耕地保育重点实验室,武汉430070)
天然有机质(Natural Organic Matter,NOM)在土壤、沉积物和水体中无处不在,存在溶解性有机质(Dissolved Organic Matter,DOM)和固体有机质两种形式。环境中腐植酸(Humic Substance,HS)是NOM的主要组成部分。地下水中HS的浓度以溶解性有机碳(Dissolved Organic Carbon,DOC)计为1~70 mg·L-1;表面水中DOC的浓度在0.5~100 mg·L-1,平均浓度5 mg·L-1;森林土壤中 HS的浓度可高达 60 g·L-1[1]。根据溶解性的差异,HS可分为富里酸(Fulvic Acid,FA)、胡敏酸(Humic Acid,HA)和胡敏素(Humin),其含有不同类型的活性官能团[如羧基(-COOH)、酚基(-OH)、氨基(NHR、-NH2)和硫醇基(-RS)]可与金属离子相互作用,改变其形态和形态分布[2-6]。与此同时,HS以及HS与土壤矿物的结合影响矿物与金属离子的反应特性,在控制环境中金属离子的迁移性、生物有效性和毒性等方面起着重要作用[7-11]。
在矿物表面上存在有机包裹层,即便是不均匀的,也会引起表面空间和静电变化,从而改变胶体矿物颗粒的物理化学性质,如表面电荷和胶体稳定性,进而改变金属氧(氢氧)化物矿物的吸附能力[1,11]。矿物-有机混合体系中二者间相互作用可能会导致对金属吸附的拮抗或协同效应,这取决于金属离子的类型和浓度、体系pH以及矿物和有机物的表面化学性质[7]。二价阳离子的存在通常会增加矿物对天然有机物质的吸附,然而NOM对金属离子吸附的影响跟pH有关[12]。与金属离子-氧化物二元体系相比,三元体系中的一般情况是:在低pH且存在NOM情况下,矿物表面金属离子的吸附增强[12]。
金属离子与矿物表面吸附的有机质之间的相互作用尚不完全清楚,已有的报道也存在争议。可能的机制包括:(1)NOM和金属离子竞争表面活性位点;(2)在溶液中形成离子-NOM络合物,减少了金属离子在矿物表面的吸附;(3)形成三元表面络合物;(4)静电作用可能改变表面电荷及表面电荷分布[12];(5)在矿物表面吸附期间,发生不同大小分子量的NOM分级[7]。其中,三元表面络合物一般可以形成两种形式:(1)金属离子桥接矿物表面位点与NOM形成A型三元络合物;(2)NOM与矿物表面结合,而金属阳离子与NOM分子中未与矿物表面位点结合的官能团结合形成B型三元络合物[12]。
本文系统总结了FA/HA等NOM和Cu(Ⅱ)、Ni(Ⅱ)、Pb(Ⅱ)、Cd(Ⅱ)、Zn(Ⅱ)等重金属离子在铁/铝氧化物、方解石、白云母及高岭石等矿物表面的共吸附特性及机制。也简要讨论了NOM和镅[Am(Ⅲ)]、铕[Eu(Ⅲ)]、钕[Nd(Ⅲ)]、钚[Pu(Ⅳ)]、钍[Th(Ⅳ)]及铀[U(Ⅵ)]等稀土或放射性金属离子在矿物表面的相互作用。除了宏观溶液化学实验,表面络合模型和光谱技术(如X射线吸收光谱)的运用促进了对微观共吸附机制的认识。这有利于系统认识NOM存在条件下重金属离子在矿物-水界面迁移与固定过程和机制,也有助于指导复杂环境中重金属元素的污染修复。
1 天然有机质和常见金属离子在矿物表面的共吸附
1.1 宏观溶液化学研究
采用宏观溶液化学方法可研究NOM和金属离子在矿物表面的共吸附特性和过程。吸附研究表明,NOM一般可促进矿物对金属离子的吸附容量,但对不同离子的影响不同[13-16]。经过HA包裹后的纳米Fe3O4颗粒对Cu(Ⅱ)、Cd(Ⅱ)的最大吸附量分别增加了231.5%和75.8%。纯纳米Fe3O4对3种重金属离子的吸附能力的强弱为Pb(Ⅱ)>Cd(Ⅱ)>Cu(Ⅱ),而经HA包裹后的顺序则为Pb(Ⅱ)>Cu(Ⅱ)>Cd(Ⅱ)。与纯纳米Fe3O4相比,经HA包裹的纳米Fe3O4对Cu(Ⅱ)和Cd(Ⅱ)具有较高的吸附量和吸附亲和力参数,而对Pb(Ⅱ)的吸附无显著性差异[13]。HA可促进纳米Fe3O4颗粒对重金属离子Hg(Ⅱ)、Pb(Ⅱ)、Cd(Ⅱ)和Cu(Ⅱ)的吸附,最大吸附容量为46.3~97.7 mg·g-1。在最优pH条件下,Fe3O4/HA能够去除天然水和自来水中99%以上的Hg(Ⅱ)和Pb(Ⅱ)以及95%以上的Cu(Ⅱ)和Cd(Ⅱ)[14]。与单一赤铁矿对Cd(Ⅱ)的吸附相比,由于带负电荷的HA与带正电荷的赤铁矿之间的静电作用,多元体系对Cd(Ⅱ)的吸附量增加[16]。氧化铝纳米颗粒、HA和Zn/Cd三元体系中可能是由于形成了三元络合物,HA增强了矿物对Zn和Cd的吸附[17]。类似地,高岭石-HA复合物对Cu(Ⅱ)的吸附量明显大于纯高岭石。这是由于HA含有大量的羧基和酚羟基等活性基团,吸附在高岭石上的HA增加了其表面吸附位,形成了高岭石-HA-Cu三元配合物,且Cu(Ⅱ)的吸附量与复合物中HA的含量在一定范围内呈正相关[18]。FA和HA对Mg-Al层状双氢氧化物(LDH)去除Ni(Ⅱ)具有很强的协同作用,在pH 7时对Ni(Ⅱ)的吸附量最高分别可达(290.6±16.4)mg·g-1和(480.4±21.3)mg·g-1,显著高于单一 LDH对Ni(Ⅱ)的吸附量(204.2±10.6)mg·g-1。Ni(Ⅱ)掺入到Mg-Al LDH结构中形成Ni-Al LDH并释放出Mg(Ⅱ),另外结合在LDH表面的HA或FA可络合Ni(Ⅱ),从而导致对Ni的去除效果增强[19]。然而,NOM对金属离子吸附的影响,也存在一些特殊情况,例如溶解性腐殖质对磷灰石吸附Cd的影响较小,但影响所吸附Cd(Ⅱ)的解吸[20]。
同时,金属离子一般也影响矿物对NOM的吸附。酸性pH条件下,Pb(Ⅱ)的存在增加针铁矿对HA的吸附[21]。pH影响HS在针铁矿上的吸附,针铁矿表面结合的Pb促进HS的吸附[3]。在弱酸性pH下,Cu(Ⅱ)的存在降低了DOM在针铁矿表面的吸附,但在高pH下DOM的吸附增强;在施氏矿物、Cu(Ⅱ)和DOM三元体系中,Cu(Ⅱ)不影响DOM的吸收[22]。可见,金属离子对NOM在矿物表面的吸附也与体系pH、矿物类型和金属离子的类别有关。
NOM和金属离子共吸附体系中,pH影响矿物对金属离子的吸附特性。一般,低pH下NOM促进矿物对金属离子吸附,而高pH下则降低金属离子吸附。有研究发现,在低pH时,FA分别促进赤铁矿、勃姆石、高岭石、钠基累托石和膨润土对Cu(Ⅱ)[23-24]、Ni(Ⅱ)[8]、Pb(Ⅱ)[25]和 Co(Ⅱ)[26]的吸附,而在高 pH 时表现出抑制作用。高pH时,在水相中形成溶解性的金属离子-NOM络合物的浓度增加[23-24]。此外,适度低pH条件下,DOM增强了针铁矿体系中Cu(Ⅱ)的吸附,但对施氏矿物体系没有影响。可见,三元体系中DOM与金属离子的吸附行为也受矿物类型的影响。
NOM和金属离子的添加顺序影响矿物对金属离子的吸附行为。在较高Cd(Ⅱ)浓度时,HA和Cd(Ⅱ)的添加顺序对赤铁矿吸附Cd(Ⅱ)有显著的影响,对Cd(Ⅱ)吸附的趋势是:在HA之前添加Cd(Ⅱ)>同时添加HA和Cd(Ⅱ)≈在HA之后添加Cd(Ⅱ)[27]。HA和Cu(Ⅱ)溶液按先后顺序或同时加入高岭石中,在Cu(Ⅱ)平衡浓度<10 mg·L-1时,3种加入顺序对Cu(Ⅱ)的吸附量基本相同;当Cu(Ⅱ)平衡浓度>10 mg·L-1时,在HA之前添加Cu(Ⅱ)和同时添加HA和Cu(Ⅱ)对Cu(Ⅱ)的吸附量比在HA之后添加Cu(Ⅱ)的略大[18]。此外,FA和Co(Ⅱ)的添加顺序影响膨润土对Co(Ⅱ)的吸附[26]。这说明NOM和金属离子在矿物表面的共吸附特性取决于其添加顺序,并且不同添加顺序下的反应机理存在差异。
矿物、NOM和金属离子三元体系中的共存离子或配体也影响着金属离子的吸附。EDTA络合作用显著降低了HA/赤铁矿体系中Cd(Ⅱ)的吸附[27]。海水体系中Hg(Ⅱ)在氢氧化铁和氢氧化锰表面的吸附受到抑制,是因为在溶液中形成稳定的HgCl2。HA的加入抑制了淡水体系中Hg(Ⅱ)在氢氧化铁和氢氧化锰表面的吸附,这可能是由于Hg(Ⅱ)和HA在矿物表面的竞争吸附造成的;与此相反,HA的加入促进了海水体系中氢氧化铁和氢氧化锰对Hg(Ⅱ)的吸附,这可能是由于加入HA导致Cl-和HA之间发生反应,因此促进了矿物对Hg(Ⅱ)的吸附[28]。
总之,宏观吸附研究表明,NOM通常促进矿物对金属离子的吸附。矿物、NOM和金属离子三元体系中,pH、矿物类型、金属离子和NOM的类别、共存的其他离子或配体、NOM和金属离子的添加顺序等均会影响矿物对金属离子的吸附特性。一般宏观溶液化学研究可了解金属离子和NOM的基本吸附特性和历程,而结合表面络合模型和光谱技术可进一步深入认识其作用机制。
1.2 表面络合模型研究
表面络合模型有三层模型(Triple Layer Model,TLM)、非理想竞争吸附-杜南(Non-ideal Compective Adsorption-Donnan,NICA-Donnan)模型、电荷分布-多位点络合(Charge Distribution-Multi Site Complexiation,CD-MUSIC)模型和配位电荷分配(Ligand and Charge Distribution,LCD)模型等,这些模型不仅能定量描述矿物和NOM对金属离子的吸附行为,揭示吸附机制,还可计算金属离子的形态分布,预测它们在环境中的化学行为[29]。其中,TLM模型考虑各种离子电荷对静电势的影响,双电层具有C1、C2两个电容值;电解质离子对矿物表面电荷也有一定贡献,可与表面位点形成离子对化合物。当溶液仅存惰性电解质离子时,矿物表面反应主要是电解质离子专性吸附和质子解离反应。CD-MUSIC模型考虑矿物表面吸附位点的多样性和异质性,静电双电层中被吸附离子的空间分布,所带电荷对静电面的电荷贡献和对静电势的影响,还包括了离子与表面位点间的络合反应。CDMUSIC模型的参数包括位点密度、比表面积、Stern层电容、质子亲和常数、离子对亲和常数以及平均位点密度等。用于描述和预测矿物(金属氧化物)对质子、金属离子、含氧阴离子、小分子有机物以及FA的吸附。NICA-Donnan模型假设HS具有低亲和性(羧基类)和高亲和性(羟基类)两类酸性位点,位点的质子亲和常数连续分布[29]。NICA-Donnan模型可用于描述HS对质子、金属离子的吸附:对质子吸附的参数有位点密度(Qmax,H1,Qmax,H2)、质子亲和常数(logKH1,log-KH2)、表观化学异质性参数(mH1,mH2)和静电常数(b);对金属离子(Men+)吸附的参数包括亲和常数(logKMe1,logKMe2)、非理想吸附参数(nH1,nH2,nMe1,nMe2)和内在化学异质性参数(p1,p2)。LCD模型是以NICA-Donnan模型和CD-MUSIC模型为基础提出和发展的化学形态模型,假设HS的羧基与矿物表面位点形成内圈复合物,可用于描述矿物对有机小分子、HS的吸附,以及矿物-HS复合物对金属离子或含氧阴离子的吸附[29]。
宏观吸附研究与表面模型结合,有助于深入认识三元体系中NOM和金属离子的配位机制。一般在较低pH时,NOM促进矿物对金属离子的吸附,相关研究如表1所示。CD-MUSIC模型研究表明,相对于二元体系,由于HA吸附到针铁矿表面后,质子竞争HA官能团的能力下降以及针铁矿-水界面静电势的变化,三元体系中Cu(Ⅱ)的吸附增强[30]。相对于二元体系,在pH 4和pH 6时,三水铝石、HA和Cu(Ⅱ)三元体系对Cu(Ⅱ)的吸附量更大。CD-MUSIC和NICA-Donnan模型研究表明,在pH 4时,单独三水铝石对Cu(Ⅱ)的吸附量非常小,但存在HA时可能是由于形成三元络合物以及由于它们相互作用导致矿物表面或HA处的静电势的变化,三水铝石对Cu(Ⅱ)的吸附显著增加[31]。在低pH条件下FA促进γ-Al2O3表面对Cu(Ⅱ)和Pb(Ⅱ)的吸附。TLM模型模拟结果表明,在溶液中和在γ-Al2O3表面,Cu(Ⅱ)和Pb(Ⅱ)均主要与FA结合[32]。HS的存在促进针铁矿-HS络合物对Pb(Ⅱ)的吸附。CD-MUSIC和NICA-Donnan模型分析表明,在低pH值时,由于针铁矿吸附的HS能够强烈地降低Stern层和Stern面的静电势,所以针铁矿-HS复合物结合Pb(Ⅱ)的量增加[3]。
模型分析表明,三元体系中NOM和金属离子的吸附特性和机制受pH、离子强度、金属离子和NOM浓度的影响。使用A型(针铁矿-Cu-FA)和B型(针铁矿-FA-Cu)三元络合物,LCD模型对Cu(Ⅱ)吸附的描述与实验数据吻合。在低pH、低浓度Cu(Ⅱ)和高浓度FA情况下,阳离子桥接的A型三元络合物是主要的Cu形态;而在高pH、高浓度Cu(Ⅱ)和低浓度FA下,Cu仅与针铁矿结合是主要的吸附机制。Cu(Ⅱ)与吸附的FA配体结合形成B型三元络合物不是Cu(Ⅱ)吸附的主要机制[12]。随着HA-针铁矿体系中pH和HA浓度的增加,Cu(Ⅱ)的吸附增加,天然有机质-电荷分布模型(NOM-CD)计算可以描述并预测Cu(Ⅱ)的反应性[33]。在pH 6以下,与纯赤铁矿相比,存在FA时赤铁矿对Cu(Ⅱ)的吸附增加40%。在低离子强度(0.01 mol·L-1)下,FA对Cu(Ⅱ)在赤铁矿表面吸附的影响比在高离子强度(0.1 mol·L-1)下更显著。实验数据与NICA-Donnan模型计算的结果相比较,表明使用线性叠加假设赤铁矿、Cu(Ⅱ)和FA三元体系中的Cu(Ⅱ)吸附低被估了30%[23]。可见,NOM对Cu(Ⅱ)吸附的影响与反应体系有关。此外,双层模型(DLM)计算发现在高pH值下,NOM会抑制针铁矿表面对金属离子Cu(Ⅱ)、Cd(Ⅱ)和Ni(Ⅱ)的吸附,天然有机配体没有形成三元表面络合物[34]。与纯针铁矿对Ca(Ⅱ)的吸附相比,存在FA时矿物对Ca(Ⅱ)的吸附显著增强。与FA和针铁矿结合Ca(Ⅱ)的线性叠加量相比,在低pH下针铁矿和FA之间的相互作用导致对Ca(Ⅱ)吸附减少,而在高pH下吸附增强。LCD模型计算表明,在针铁矿表面Ca(Ⅱ)和FA之间的相互作用主要是静电作用[35]。LCD模型分析表明小分子FA在针铁矿-水界面的Stern层中均匀分布,而HA大颗粒分布于Stern层和扩散层[36]。低浓度Pb(Ⅱ)时,几乎所有的Pb(Ⅱ)均桥接FA/HA与针铁矿。高浓度Pb(Ⅱ)和低浓度HS时,在所研究的pH范围内,针铁矿-Pb络合物占主导地位;但在高浓度HS,低pH时主要的络合物形态是针铁矿-HS-Pb三元络合物(B型),而在高pH时是针铁矿-Pb络合物。可见,针铁矿、HS和Pb(Ⅱ)三元体系的共吸附机制不同于针铁矿、FA和Cu(Ⅱ)体系[12,36],后者形成A型三元络合物,表明近似条件下的吸附机制与金属离子的类型有关。
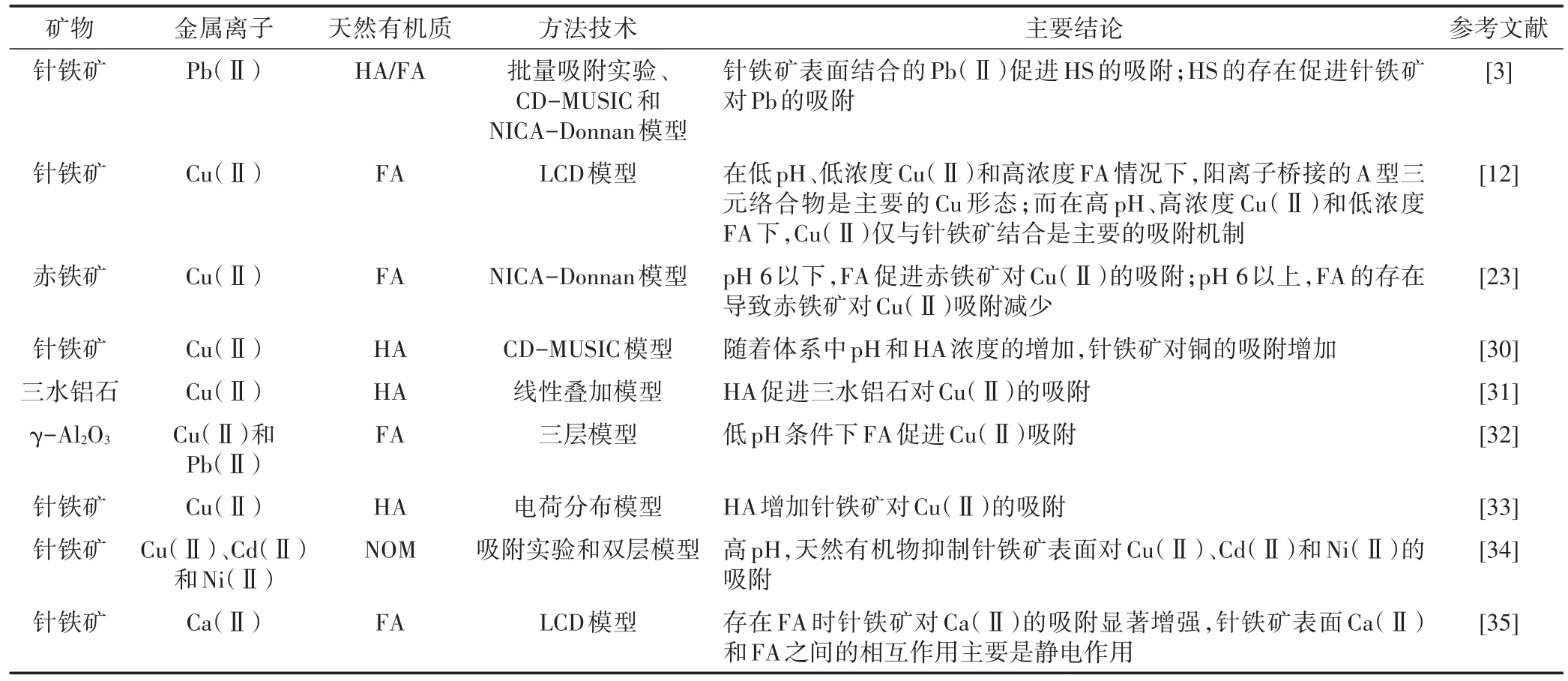
表1 天然有机质和金属离子在矿物表面的共吸附(表面络合模型研究)Table 1 Co-sorption of natural organic matter and metals on minerals(studied by surface complexation modeling)
表面络合模型研究可揭示三元体系中NOM和金属离子的配位机制,预测金属离子的形态和移动性;金属离子吸附机制受NOM及金属离子浓度、金属离子类型、体系pH及离子强度等因素的影响。目前,表面络合模型主要用于研究针铁矿、赤铁矿、三水铝石等体系,且限于Cu(Ⅱ)、Pb(Ⅱ)等少数元素,其他矿物、金属离子和NOM三元体系的应用还有待进一步加强。
1.3 X射线吸收精细结构光谱研究
X射线吸收精细结构光谱(XAFS)包括X射线吸收近边结构(XANES)和扩展边X射线吸收精细结构(EXAFS)。采用XAFS光谱技术有助于进一步深入分析三元体系中金属离子与近邻原子配位的种类、数量和距离等结构信息,进而从分子水平理解金属离子的共吸附机制,相关研究总结见表2。HA增强了高岭石对Zn的吸附,EXAFS分析表明,在HA存在及不存在的情况下,Zn-O的配位距离为1.91~2.02 Å,Zn-Al的配位距离为3.11 Å,Zn在高岭石表面均形成共边的双齿内圈络合物[37]。在pH值为8.25时,方解石对Cu(Ⅱ)的吸附随着溶解性HA浓度的增加而降低,表明形成Cu-HA水溶性络合物是控制方解石表面对Cu(Ⅱ)吸附的主要因素。XAFS光谱分析显示,存在和不存在HA的情况下,Cu-C的原子间距为2.83~2.95 Å,Cu-Ca的原子间距为 3.85~3.90 Å,表明方解石表面吸附Cu(Ⅱ)的局部配位非常相似。然而,二元体系吸附样品和三元体系吸附样品之间XAFS光谱的细微差异表明不能排除形成三元表面络合物的可能。因此,方解石表面结合的HA对Cu(Ⅱ)吸附的影响很小,表明Cu(Ⅱ)与方解石表面位点配位,而不是与表面结合的HA配位[38]。XAFS光谱分析显示,在较低HA浓度时,相对于针铁矿-Cu(Ⅱ)和HA-Cu(Ⅱ)体系,三元共吸附体系Cu-O键长有所减小,为2.24~2.27 Å,由此判定Cu(Ⅱ)与针铁矿及HA配位主要形成A型三元络合物,即Cu(Ⅱ)与针铁矿(001)表面结合形成内圈络合物,再与HA的羧基或酚基配位;而在较高HA浓度时,根据Cu-O和Cu-Fe键长特点判定形成B型三元络合物,即Cu(Ⅱ)与HA结合,而HA与针铁矿表面结合。针铁矿吸附较高浓度HA使得Cu(Ⅱ)不易接近矿物表面位点[39]。然而,EXAFS光谱分析表明,勃姆石-Ni(Ⅱ)-FA三元体系中,较低pH和较高FA浓度时形成B型三元表面络合物[勃姆石-FA-Ni(Ⅱ)],而在较高pH和低FA浓度时Ni(Ⅱ)直接与勃姆石表面络合,即形成勃姆石-Ni(Ⅱ)二元络合物或Ni桥接的A型三元络合物[勃姆石-Ni(Ⅱ)-FA]。不同形态络合物的相对贡献在很大程度上取决于溶液化学条件;B型三元络合物的浓度随着FA吸附量的增加和pH的降低而增加[8]。类似地,HA存在时,伊利石对Ni(Ⅱ)的吸附机制受pH和老化时间的控制。在中性至弱碱性条件下及较短的老化时间内,HA抑制了Ni(Ⅱ)在伊利石表面的吸附。随着老化时间的延长,HA的抑制作用变弱。在低pH条件下,离子交换和形成B型三元络合物是主要的吸附机制。HA对Ni(Ⅱ)吸附的抑制作用可能是由于形成了可溶性的HA-Ni络合物或HA包裹在伊利石表面抑制了表面诱导沉淀物的形成[40]。可见,不同pH、NOM浓度条件下,反应机制因体系而异。NOM存在条件下,金属离子在矿物表面形成内圈络合物或在矿物表面形成三元络合物是其主要的反应机制。
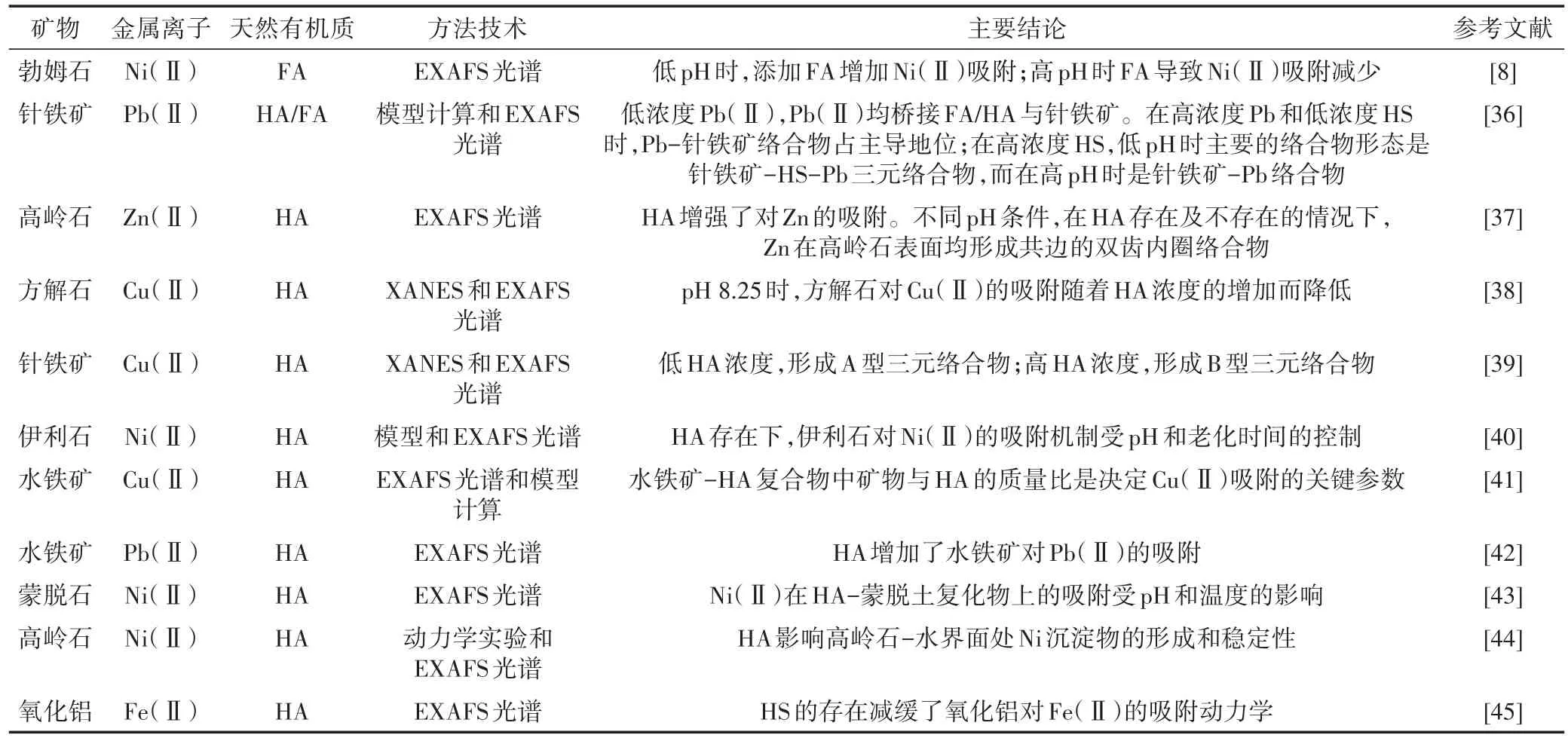
表2 天然有机质和金属离子在矿物表面的共吸附(X射线吸收精细结构光谱技术研究)Table 2 Co-sorption of natural organic matter and metals on minerals(studied by XAFS)
矿物表面NOM的负载量影响金属离子在矿物-NOM组分上的分布。使用HS结合态Pb和针铁矿吸附态Pb作为参考物,基于EXAFS的线性拟合表明,随着HS浓度的增加,更多的Pb(Ⅱ)与吸附的HS结合,而与针铁矿的结合减少,这与LCD模型计算吻合[36]。表面络合模型和Cu-K边EXAFS分析表明,对于水铁矿-HA复合物,其中矿物与HA的质量比是决定Cu(Ⅱ)吸附的关键参数,羧基与Cu(Ⅱ)的结合是控制Cu(Ⅱ)归属与迁移的关键[41]。与通过预吸附HA形成的水铁矿等同复合物相比,通过共沉淀形成的复合物具有较高的碳含量、较小的比表面积和较快的Pb(Ⅱ)吸附速率[42]。在pH <5时,复合物对Pb(Ⅱ)吸附比纯水铁矿高,并且共沉淀和预吸附HA的复合物吸附几乎等量的Pb(Ⅱ)。Pb-LⅢ边EXAFS光谱分析表明Pb(Ⅱ)在水铁矿表面上形成双齿共边Pb络合物,其中Pb-Fe原子间距为3.33 Å;Pb(Ⅱ)与水铁矿-HA复合物中的HA配位形成双齿内圈Pb络合物,Pb-C的距离约为3.1 Å。随着pH从4增加到6.5,更多的Pb(Ⅱ)与复合物的HA结合。预吸附HA的有机部分比在共沉淀的复合物吸附更多的Pb(Ⅱ)[42]。
天然有机质影响金属离子Ni(Ⅱ)/Fe(Ⅱ)等在粘土矿物和铝氧化物表面的吸附-沉淀转化过程,EXAFS光谱有助于认识其反应机制。Ni(Ⅱ)在蒙脱土-HA复合物上的吸附受pH和温度的影响。EXAFS光谱表明,在低pH条件下,Ni(Ⅱ)的吸附主要以蒙脱土-HA-Ni三元络合物和外圈络合为主。在高pH值时,二元蒙脱土-Ni表面络合是主要的吸附机制。pH 10时,溶解性HA-Ni(Ⅱ)络合物抑制了Ni(OH)2(s)的形成,而形成Ni-Al LDH相[43]。HA负载影响高岭石-水界面处Ni沉淀物的形成和稳定性。初始Ni(Ⅱ)吸收量随着高岭石表面所负载HA量的增加而增大。EXAFS光谱分析显示,HA负载1%(wt)时,Ni(Ⅱ)在高岭石表面形成Ni-Al LDH沉淀,而负载5%(wt)时,表面沉淀的形成显著减慢,并且形成与Ni(OH)2(s)结构类似的沉淀[44]。EXAFS光谱分析表明,Fe(Ⅱ)-Al(Ⅲ)LDH沉淀是Fe(Ⅱ)吸附的主要形态,HS的存在降低了氧化铝对Fe(Ⅱ)的吸附速率,HS与溶出的Al形成络合物也减缓了氧化铝悬浮液中Fe(Ⅱ)的沉淀,但不影响形成Fe(Ⅱ)-Al(Ⅲ)LDH的组成和稳定性;黏土体系中,HA与Al的络合以及黏土悬浮液中的溶解性硅使得形成层状硅酸盐沉淀,导致主要的Fe(Ⅱ)吸附产物从Fe(Ⅱ)-Al(Ⅲ)LDH变为含有少量结构Al的Fe(Ⅱ)-层状硅酸盐,Fe(Ⅱ)沉淀机制的变化并不影响较低HA水平下Fe(Ⅱ)的吸附速率[45]。此外,EXAFS光谱技术也有助于分析土壤体系中Ni的吸附与沉淀转化。土壤环境中,在短时间内,Ni(Ⅱ)主要吸附在SOM上,而经过长期反应后形成Ni-LDH沉淀是导致Ni(Ⅱ)固存的原因。经过较长的反应时间,吸附的Ni可以缓慢地转化成Ni-LDH沉淀[46]。
总之,基于同步辐射的XAFS光谱分析有助于在分子尺度上进一步深入分析三元体系中金属离子的配位机制,以及可能存在的沉淀反应。吸附机制同样受矿物类型、NOM及金属离子浓度、金属离子类型、体系pH、老化时间等因素的影响。
1.4 其他新型技术研究
新型研究技术,如长周期X射线驻波荧光光谱(LP-XSW-FY)技术、X射线反射率技术(XR)等丰富了对矿物-NOM-金属离子三元体系中金属离子吸附机制的研究。LP-XSW-FY可探测凝固相或液相样品中金属原子层的空间纵深结构[1];XR是原位非破坏性分析技术,适合于探测矿物-水界面溶解性有机质和金属阳离子的吸附与分布[47]。LP-XSW-FY分析表明,HA和金属氧化物表面的固有特性,如结合位点的性质、结合亲和力和表面电荷等,以及pH、共存竞争离子等介质条件是控制金属离子在HA和金属氧化物表面分布和形态的关键因素;HA存在时,Pb(Ⅱ)在α-Al2O3(1-102)和α-Fe2O3(0001)活性面上吸附发生了再分布;随着反应时间的增加,Cu(Ⅱ)和Zn(Ⅱ)从HA吸附层到α-Fe2O3(0001)面的分配略微增加,而在α-Al2O3(1-102)表面上的分配没有发生变化,表明这些离子与矿物表面吸附HA的官能团强烈络合[1]。通过XR研究FA对Cu(Ⅱ)、Zn(Ⅱ)和Pb(Ⅱ)等离子在白云母(001)表面上的吸附表明,吸附模式受FA-阳离子结合强度和阳离子水合焓控制,对于弱水合阳离子,FA的存在增加约60%~140%的金属离子吸附;Cu(Ⅱ)和Zn(Ⅱ)水合强度更强,在白云母表面上的吸附呈现两种不同的外圈络合物,与无FA条件下的分布相比变化很小,表明尽管它们对FA具有较强的亲和力,但强烈的水合作用阻碍了它们与FA的进一步结合[47]。可见,新型光谱技术对于金属离子和NOM在矿物表面的共吸附可提供原位结构信息和新的认识。
2 天然有机质和稀土、放射性金属离子在矿物表面的共吸附
由于稀土的大量开采和应用,稀土元素进入土壤环境的数量急剧上升。低浓度的稀土元素对农作物生长发育有促进作用,但高浓度喷施量则对作物的根和地上部分有抑制作用。稀土元素在土壤环境中可以累积,并成为污染物[48]。另外,从核能开发到应用过程中所产生的放射性元素是环境的主要放射性污染来源。放射性元素能长期存在于土壤和水体中,在水体中具有高度可溶性和流动性,可直接或间接地进入到食物链中,对生态系统及人类健康造成严重威胁[49]。稀土、放射性金属离子在矿物表面的吸附影响其流动性和生物有效性。近年来,NOM和稀土元素、放射性元素在矿物表面的共吸附也广受关注。
pH是影响NOM和稀土、放射性金属离子在矿物表面共吸附的重要因素之一。在低pH时,HA的存在增加α-Al2O3对Eu(Ⅲ)的吸附,而在高pH时抑制矿物对Eu(Ⅲ)的吸附,同时Eu(Ⅲ)轻微增加α-Al2O3对HA的吸附[50-51]。低pH时FA促进了Eu(Ⅲ)在二氧化硅表面的吸附,但在高pH时降低Eu(Ⅲ)的吸附,并形成了两种三元表面络合物,其中Eu(Ⅲ)桥接矿物表面位点和FA[52]。在针铁矿-Nd(Ⅲ)-FA三元体系中,pH等于针铁矿电荷零点(PZC)时,FA增强Nd(Ⅲ)的吸附,这是由于形成B型三元表面络合物(针铁矿-FA-Nd);pH低于针铁矿PZC时,Nd(Ⅲ)-FA络合促进Nd(Ⅲ)吸附;pH高于PZC时,特别是在FA浓度升高的情况下,Nd(Ⅲ)-FA络合物的形成抑制了Nd(Ⅲ)的吸附[53]。pH 4.6时,铀酰基[U(Ⅵ)O22+]-水铁矿-HS三元体系中,HA的存在比FA更能增强水铁矿对铀酰基的吸收;在pH 7时,HA和FA(0~500 mg·L-1)都不影响水铁矿对铀酰吸附。在最高HS浓度(500~955 mg·L-1)下,由于水铁矿聚集体发生分散,对铀酰基的吸附在pH 7时受到轻微抑制[54]。相对于针铁矿-Pu(Ⅳ)体系,在pH 5和pH 7时,三元体系中由于FA和HA与Pu(Ⅳ)的络合作用,降低了针铁矿对Pu(Ⅳ)的吸附;相反,在pH 3时FA和HA增加了针铁矿对Pu(Ⅳ)的吸附,表明形成三元络合物或HA存在下结合到其聚集体中[55]。
NOM、反应时间和加入顺序影响稀土元素、放射性元素在矿物表面吸附及解吸的速率和程度。利用阳离子交换树脂研究了HA-γ-Al2O3所结合243Am(Ⅲ)的解离动力学,结果表明放射性核素243Am(Ⅲ)与HA包覆的Al2O3的不可逆键合部分随着pH增加而增加,并且不依赖于老化时间。随着老化时间的增加,HAAl2O3胶体中存在的243Am(Ⅲ)从“快”离解位点转移到“慢”离解位点[56]。此外,Bouby等研究了天然地下水中锕系重金属离子Cs(Ⅰ)、Eu(Ⅲ)、Th(Ⅳ)和U(Ⅵ)与蒙脱石胶体的相互作用。对于Eu(Ⅲ)体系,即便预先老化大约3年后加入竞争配体HA,也会导致胶体所吸附的金属离子快速解离。而在Th(Ⅳ)体系中仅发生部分解离。实验和计算表明,HA络合物控制所有金属离子的形态[57]。在pH 4时,FA改变针铁矿对Pu(Ⅳ)的吸附速率和程度[58]。FA存在情况下Pu(Ⅳ)的解吸比例下降,表明有机质可以稳定吸附在针铁矿表面的Pu(Ⅳ)。三元Pu-FA-矿物络合物可以增强胶体促进的Pu(Ⅳ)迁移[58]。Th(Ⅳ)在铁氧化物上的吸附取决于HA与铁氧化物表面位点的比例[59]。当HA和赤铁矿预先平衡24 h,赤铁矿对Th(Ⅳ)的吸附受到影响;当在赤铁矿-Th(Ⅳ)体系平衡24 h之后添加HA,Th(Ⅳ)几乎不从赤铁矿表面上解吸;HA浓度的增加仅导致Th(Ⅳ)吸附的轻微单调下降。表明HA和表面位点间的比例对Th(Ⅳ)吸附没有显著影响,体系组分间的接触时间增加只引起Th(Ⅳ)吸附的轻微变化[60]。
总之,包括稀土、放射性元素在内的金属离子和HA/FA等天然有机质在矿物表面的共吸附行为和机制,受金属离子和矿物的性质以及溶液化学性质,如吸附剂浓度、pH和温度等多重因素的影响。现代化的光谱技术结合机制模型计算有助于深入认识金属离子-天然有机质多组分在矿物界面的共吸附特性及分子机制。
3 展望
天然有机质和金属离子(包括稀土金属元素、放射性元素等)在矿物表面的共吸附行为和机制,受金属离子和矿物的性质以及溶液化学条件,如吸附剂浓度、pH和温度等多因素的影响。笔者认为,进一步系统和深入认识天然有机质和金属离子在矿物表面的共吸附行为和机制还需从以下几个方面加强研究:
(1)为认识真实环境中天然有机质-金属离子在矿物表面吸附行为,需结合实验室研究,对比探讨场地或田间等真实环境中,天然有机质对金属离子在矿物表面吸附的影响。
(2)进一步探讨多矿物组分条件下,天然有机质对金属离子在矿物表面吸附的机制,可阐明复杂环境中天然有机质-金属离子的交互作用。
(3)为深入探讨天然有机质-金属离子在矿物表面共吸附行为,仍需要研究不同分子大小和来源的天然有机质对金属离子在矿物表面选择性吸附的影响及其规律性。
(4)发展新的分子水平的表征方法研究天然有机质与金属离子在矿物表面的共吸附机制,以便开发更为有利的、可靠的模型以预测毒性金属离子在环境中的迁移与归宿。同时也需进一步研究以探讨模型参数的适合性及一般性。
