CRISPR/Cas9介导的小鼠BRCA1/BRCA2/CDH1基因乳腺特异性修饰研究
2018-08-30杨漫漫张婷婷陈文彬
陈 茜,向 熙,杨漫漫,4,张婷婷,陈文彬,李 林,魏 强,4,李 勇,,4*
(1.深圳华大方舟生物技术有限公司,深圳 518000; 2.深圳华大生命科学研究院,深圳 518000; 3.中国科学院大学华大教育中心,深圳 518000; 4.深圳动物基因组辅助育种工程实验室,深圳 518000)
乳腺癌是女性常见的主要恶性肿瘤之一[1-2]。我国虽然属乳腺癌低发国家,但人口基数大,乳腺癌已然成为我国女性发病率最高的癌症,严重威胁了广大女性的生命健康[3]。BRCA1(breast cancer susceptibility gene1)是最先被发现的高共显乳腺癌易感基因。研究发现,有乳腺癌家族史的妇女中, BRCA1的基因突变比例可达15%~20%;在同时具有乳腺癌和卵巢癌家族史的妇女中,BRCA1的基因突变比例更高,甚至高达60%~80%[4]。除了BRCA1外,BRCA2(breast cancer susceptibility gene 2)和CDH1(cadherin gene 1)也是迄今为止发现重要乳腺癌易感基因。大规模乳腺癌群体的高通量测序及分析研究也证实,BRCA1、BRCA2和CDH1三个基因结构和功能的异常与乳腺癌的发病密切相关[5]。
为了探索乳腺癌的发病机制和探讨新的治疗方法,国内外研究者需借助更贴近于人类的原发性乳腺癌动物模型,而制备BRCA1/BRCA2/CDH1基因乳腺特异性修饰小鼠是建立该模型的有效途径。虽然基于基因敲除的小鼠模型在肿瘤发病机制研究中有独特的优势,但部分肿瘤驱动基因在动物体内的全身性纯合敲除具有胚胎致死性,很难进行后续的机理药理研究,典型的如BRCA1和BRCA2[6]。对于这些致死性基因的失活,多采用组织特异性敲除的方法,其原理是利用同源重组的方法在敲除区域两侧插入loxp片段(floxp),随后floxp小鼠与组织特异性表达Cre重组酶的小鼠杂交,Cre重组酶可识别并折叠剪切loxp中间的片段,达到组织特异性敲除特定基因的目的[7]。但该方法步骤繁琐,失败率高,周期长,不利于科研的开展。利用组织特异性启动子启动CAS9蛋白的表达,并结合全身性single guide RNA的表达,能在动物个体的特定组织或细胞群中结合成CRISPR/Cas9 RNP (ribonucleoproteins)复合物,实现靶基因的组织特异性编辑。该方法大大缩短了组织特异性敲除动物的制备周期和难度,已在斑马鱼[8]、果蝇[9]、小鼠[10]等模式动物中得到很好的应用。
本研究选取BRCA1、BRCA2和CDH1三个乳腺癌高频易感基因为靶基因位点,以CRISPR/Cas9基因编辑技术为基础,构建乳腺组织特异性的CAS9蛋白以及三条sgRNA共表达载体,通过原核注射的方式将载体序列整合到小鼠基因组中,制备转基因小鼠,使整个表达框在个体中稳定表达,从而实现乳腺组织特异性的多基因敲除。与传统的同源重组及Cre-loxp的方法相比,本实验的周期与成本可以缩短至三分之一,方便快捷地构建出乳腺组织特异性敲除小鼠,对其进行深入研究将对遗传性乳腺癌的发生机制、早期诊断及治疗有着重要的理论意义和实际应用价值。
1 材料和方法
1.1 实验动物
SPF级C57BL/6小鼠,体重25~30 g,7~8周龄,雌性30只,雄性5只。购于中国医学科学院医学实验动物研究所[SCXK(京)2013-0002],转基因动物的构建及饲养在中国医学科学院医学实验动物研究所进行[SYXK(京)2013-0014]。伦理审查号:BGI-IRB17213。
1.2 主要材料
HEPA细胞(小鼠肝癌细胞)购自上海研域生物工程有限公司;CRR质粒(可单独表达sgRNA的载体,由本实验室设计并合成);pCMV-spCas9质粒(可单独表达Cas9的载体,由本实验室设计并合成);pX330-U6-Chimeric_BB-CBh-hSpCas9载体购自Biovector公司。
无内毒素质粒提取试剂盒购自Omega公司;Lipofectamine2000转染试剂购自Thermo Fisher Scientific;Trizol试剂购自Invitrogen公司;反转录试剂盒购自北京全式金生物技术有限公司;premix ExTaq、T7核酸内切酶I、普通质粒提取试剂盒、DNA提取试剂盒、In-Fusion HD Cloning kits、琼脂糖凝胶DNA回收试剂盒购自大连宝生物公司;限制性内切酶均购自NEB公司。
1.3 实验方法
1.3.1 sgRNA设计及活性鉴定
针对小鼠BRCA1基因序列 (Gene ID:12189),利用sgRNA在线设计网站(http://crispr.mit.edu/)在其第10外显子设计并合成了1条特异识别靶序列DNA的sgRNA。合成的sgRNA及互补链经变性退火 (程序为:95℃ 10 min; 25℃ 30 min),形成带有粘性末端的双链核苷酸,再连入经BbsⅠ酶切回收的pX330载体骨架中,命名为pX330(B1)。BRCA2及CDH1基因位点的sgRNA则是根据文献报道各选取一条具有活性的gRNA直接合成[11-12],将BRCA2 sgRNA、CDH1 sgRNA分别连入经BsaI酶切回收的CRR载体骨架中,命名为CRR(B2)和CRR(C1)。构建好的表达载体经测序验证连接正确后,提取无内毒素质粒用于细胞转染。
pX330(B1)质粒直接转染HEPA细胞,CRR(B2)载体和CRR(C1)载体与pCMV-spCas9质粒共转染HEPA细胞(CRR载体与pCMV-spCas9质粒质量比为1∶1)。当24-well HEPA细胞汇合度达到50%~70%时进行脂质体转染,48 h后分别收集细胞并提取基因组DNA,PCR扩增sgRNA切割靶点上下游DNA片段(如图1 A),产物经T7程序变性退火后再经T7核酸内切酶I酶切。引物信息见表1,反应体系如下:
PCR 扩增体系:细胞基因组DNA 0.1 μg,premix ExTaq 10 μL,上、下游引物(10 μmol/L)各0.4 μL,灭菌蒸馏水补至20 μL。PCR反应条件:(98℃ 15 s;55℃ 30 s;72℃ 1 min)×32cycles。
T7变性程序:95℃ 10 min;95℃~85℃(-2.0℃/s);85℃~25℃(-0.3℃/s);25℃ 1 min。
T7酶切反应体系:PCR产物18 μL,NEBuffer2 2 μL,T7EI 0.2 μL。反应条件:37℃ 45 min。
1.3.2 WAP启动子的扩增
为确保Cas9基因在小鼠乳腺组织中特异性表达,根据小鼠基因组序列扩增乳腺组织特异性表达的WAP启动子,并在两端加入pX330骨架同源序列,与去除CBh启动子的pX330质粒骨架连接(KpnI和AgeI双酶切),重组质粒命名为pX330(WAP-Cas9-U6-B1)。
1.3.3 共表达载体的构建
扩增CRR(B2)载体上U6-BRCA2sgRNA表达框,连入经NotI酶切后的pX330(WAP-Cas9-U6-B1)骨架,命名为pX330(WAP-Cas9-U6-B1B2)。扩增CRR(C1)载体上U6-CDH1sgRNA表达框,连入经NotI酶切后的pX330(WAP-Cas9-U6-B1B2)骨架,通过限制性核酸内切酶SbfI酶切鉴定重组后的质粒,将终载体命名为pX330(WAP-Cas9-U6-B1B2C1)(如图1B),并回收线性化质粒,制备原核显微注射液。构建共表达载体所需引物信息如表2。

表1 sgRNA设计及活性鉴定引物Table.1 The primers for sgRNAs and identification of their activity
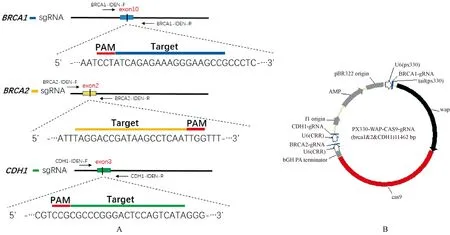
注:A:BRCA1、BRCA2和CDH1三基因sgRNA及鉴定引物设计示意图;B:共表达载体结构示意图,包含WAP启动子、U6-BRCA2-sgRNA、U6-CDH1-sgRNA。图1 BRCA1、BRCA2和CDH1三基因sgRNA及共表达载体结构示意图Note.A:Schematic diagram of sgRNAs in BRCA1、BRCA2 and CDH1 gene and the primers for identification.B:Schematic diagram of the final carrier structure, Including WAP promoter, U6-BRCA2-sgRNA, U6-CDH1-sgRNA.Figure.1 Schematic diagram of sgRNAs for BRCA1, BRCA2, and CDH1 genes and the structure of the final carrier

引物名称Primer name引物序列(5'~3')Primer sequenceWAP-infusion-FCAAATGGCTCTAGAGGTACCCTCTAAGTAGAGGGGAAGAAAWAP-infusion-RCCATGGTGGCACCGGTGGTACCGGTGTCAGGCAAGTGBRCA2-infusion-FCAGGCATGCTGGGGAGCAAGGTCGGGCAGGAAGAGGGCCTBRCA2-infusion-RCTAGGGGTTCCTGCGGCCGCAAAAAAAGCACCGACTCGGTGCCCDH1-infusion-FTCGGTGCTTTTTTTGCAAGGTCGGGCAGGAAGAGGGCCTCDH1-infusion-RAGGGGTTCCTGCGGCCAAAAAAAGCACCGACTCGGTGCC
1.3.4 转基因小鼠制备
实验第1天8:00 小鼠腹腔注射每只PMSG 7.5 IU。48 h后,实验第3天小鼠腹腔注射每只HCG 7.5 IU,注射后立即与雄鼠1∶1合笼。实验第4天8:00~9:00检查阴道栓,有阴道栓的小鼠备注日期和阳性,对阳性小鼠进行受精卵收集。向受精卵雄性原核注入显微注射液后,移植到假妊娠小鼠输卵管中,每只移入25~35枚合子。移植后将小鼠置于安静的环境下饲养,19~21 d分娩产仔。
1.3.5 转基因阳性小鼠的鉴定
剪取出生1周新生小鼠尾巴,通过试剂盒提取小鼠基因组DNA,具体提取方法按试剂盒说明书进行。根据转基因载体上的特异DNA序列进行PCR检测。PCR鉴定产物跨WAP启动子和Cas9 基因,引物见表3。PCR反应体系同1.3.1。
转基因阳性母鼠受孕分娩后,于哺乳后期采集母鼠乳腺、肾、胃、脑、脂肪和肝组织,并采集非哺乳期的母鼠乳腺、肾、胃、脑、脂肪和肝组织作为对照。Trizol法提取组织RNA后进行反转录,得到的cDNA用反转录引物扩增鉴定,引物见表3。
GAPDH引物作为内参引物,同时扩增乳腺、肾、胃、脑、脂肪和肝组织,引物序列见表3。
反转录体系:总RNA 1 μg,Oligo(dT)181 μL,2X TS Reaction Mix 10 μL,TransScript RT/RI Enzyme Mix 1 μL,RNase-free Water补至20 μL。
反转录反应条件:42℃反应30 min,85℃加热5 s 失活TransScript RT。
RT-PCR扩增体系:cDNA 2 μL,premix ExTaq 5 μL,反转录上下游引物(10 μmol/L)各0.2 μL,灭菌蒸馏水补至10 μL。
RT-PCR反应条件:(98℃ 10 s;60℃ 30 s;72℃ 20 s)×35cycles。
GAPDH引物PCR反应条件同上。
同时,试剂盒法提取这6个组织的基因组DNA,PCR分别扩增共表达载体上sgRNA切割靶点上下游DNA片段,产物经T7程序变性退火后再经T7核酸内切酶I酶切。引物信息见表4。
PCR 扩增体系:基因组DNA 0.1 μg,premix ExTaq 10 μL,上、下游引物(10 μmol/L)各0.4 μL,灭菌蒸馏水补至20 μL。
PCR反应条件:(98℃ 15 s;55℃ 30 s;72℃ 1 min)×32cycles。
T7变性程序和T7酶切反应体系见1.3.1。

表3 转基因鉴定引物及反转录鉴定引物Table.3 The primers for identifying transgenes and reverse transcription
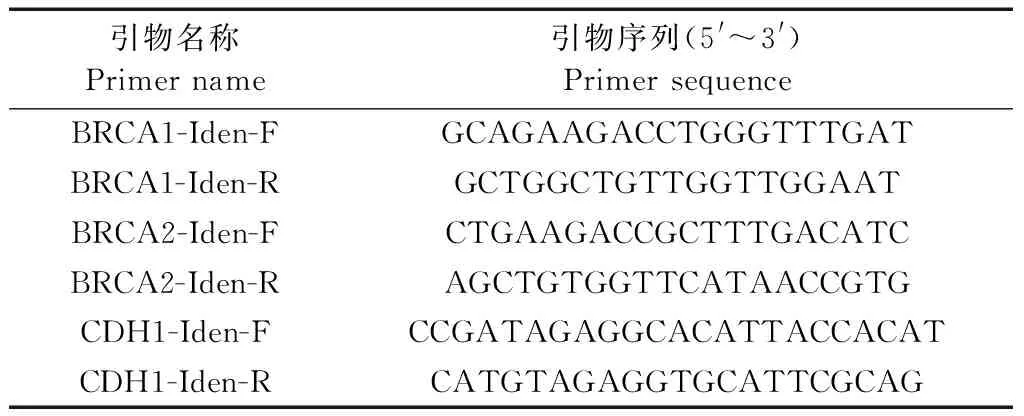
表4 共表达载体活性鉴定引物Table.4 The primers for identifying coexpression vector activity

图2 BRCA1、BRCA2及CDH1 gRNA测序验证Figure.2 gRNAs for BRCA1, BRCA2, and CDH1 sequencing
2 结果
2.1 CRISPR载体构建及活性鉴定结果
BRCA1、BRCA2和CDH1三个基因的sgRNA分别连入载体骨架后,进行测序验证,结果表明三条sgRNA均成功连入载体中(图2)。三套CRISPR系统分别转染HEPA细胞后进行sgRNA活性验证,结果显示三条sgRNA均有活性(图3),T7酶切后目的条带与预期一致:BRCA1:979 bp=440 bp+539 bp; BRCA2:408 bp=329 bp+79 bp; CDH1:731 bp=348 bp+369 bp。
2.2 共表达载体鉴定
BRCA1sgRNA连入pX330骨架,BRCA2sgRNA和U6-CDH1sgRNA整个表达框依次连入pX330(WAP-Cas9-U6-B1)酶切骨架和pX330(WAP-Cas9-U6-B1B2)酶切骨架。并成功将CBh启动子替换为WAP启动子。重组后的质粒pX330(WAP-Cas9-U6-B1B2C1)经SbfI酶切鉴定条带大小正确(图4)。
2.3 转基因小鼠的制备及转基因阳性鉴定
通过原核注射的方法制备转基因小鼠,共注射受精卵190枚,4只受体鼠移植卵共130枚,最终一共产下仔鼠42只。通过检测鉴定11只为转基因阳性鼠,阳性率为26.19%。将F0代阳性鼠与野生型小鼠交配,得到39只F1代仔鼠,经鉴定共得到9只F1代转基因阳性鼠(图5)。

注:A1:F0代小鼠PCR鉴定结果;M:200 bp marker,1~42:42只F0小鼠样品,其中11只为转基因阳性鼠;P:质粒阳性对照;N:阴性对照;B:空白对照;A2:F1代小鼠PCR鉴定结果;M:200 bp marker,1~39:39只F1小鼠样品,其中9只为转基因鼠。图5 转基因小鼠鉴定Note.A1:PCR analysis of F0 mice.M: 200 bp marker.1-42:42 Samples which include 11 transgenic mice.P: Positive control with plasmid. N: Negative control with wild mouse.B: blank control with H2O, the same below.A2: PCR analysis of F1 mice.M: 200 bp marker.1-39:39 Samples which include 9 transgenic mice.Figure.5 Identification of transgenic mice
2.4 Cas9基因表达情况鉴定
Cas9基因的表达鉴定结果显示阳性母鼠乳腺组织cDNA扩增产物大小跟预期一致,为298 bp,但胃和肝中也扩增出微弱的目的条带,见图6。同时,我们在非泌乳期母鼠乳腺、肾、胃、脑、脂肪、肝等组织中未检测到该基因的表达,这一实验结果表明WAP启动子调控的Cas9基因主要在泌乳期乳腺组织中表达。
2.5 T7酶切活性鉴定
提取阳性及野生母鼠乳腺、肾、胃、脑、脂肪、肝组织DNA,分别用鉴定引物扩增,T7酶切鉴定靶基因的剪切效果。结果显示CDH1基因有剪切活性,且仅在阳性母鼠乳腺组织发生剪切,T7酶切后目的条带与预期一致:1231 bp=570 bp+661 bp(图7)。BRCA1和BRCA2两个基因T7酶切后没有出现剪切后条带,有可能这两个基因的剪切效率较低,未能通过T7酶切方法检测出。

图3 BRCA1、BRCA2及CDH1 gRNA活性鉴定结果Figure.3 Identification of activity of three gRNAs
3 讨论
乳腺癌在世界范围内已成为女性的头号恶性肿瘤。乳腺癌的发生发展是多基因共同作用的结果,探讨它们的作用机制有助于乳腺癌预防、早期诊断及为临床治疗提供新的靶点。统计资料表明5%~10%的乳腺癌和遗传易感性相关,这其中最常见的乳腺癌易感基因是BRCA1和BRCA2,其次还有CDH1、P53、PTEN、STK11/LKB1等[13]。研究表明BRCA1和BRCA2基因的突变可以显著提高乳腺、前列腺、卵巢癌的发病率。据统计5%~10%的乳腺癌属于单基因导致的,而BRCA1和BRCA2双基因突变可以显著提高肿瘤外显性,占总体乳腺癌发病率的30%左右[14]。BRCA1和BRCA2基因与细胞内DNA损伤修复有关,它们的突变可导致基因结构不稳定,促进细胞增殖,阻止细胞正常分化,最终诱发了肿瘤的发生[15]。CDH1是一类细胞粘附相关因子,抑制肿瘤细胞的浸润和转移,该基因的突变是乳腺小叶癌的主要成因[16]。尽管有非常多关于BRCA1、BRCA2、CDH1和乳腺癌相关的研究,然而由于这些基因突变都是胚胎致死性,需要通过组织特异性的敲除才能很好的研究这些基因在乳腺癌发生、发展过程中的作用。
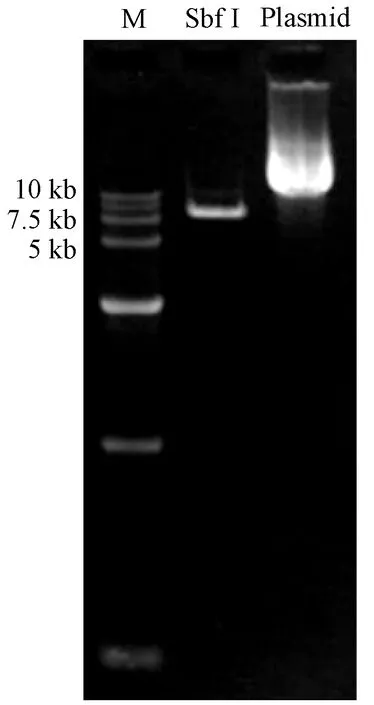
图4 pX330(WAP-Cas9-U6-B1B2C1)载体限制性内切酶酶切验证结果Figure.4 Restriction enzyme digestion identification of pX330 (WAP-Cas9-U6-B1B2C1)

注:A1:RT-Cas9-PCR引物鉴定Cas9表达结果;B:空白;M:100 bp marker;1~6:阳性母鼠乳腺、肾、胃、脑、脂肪和肝组织的cas9表达情况;A2:GAPDH扩增结果。图6 Cas9基因表达情况鉴定Note.A1:RT-Cas9-PCR to identify Cas9 expression results.B: Negative control with H2O.M: 100 bp Marker.1-6: The cas9 expression of breast tissue, kidney, stomach, brain, fat, liver in positive mice.A2:GAPDH PCR results.Figure.6 Identification of Cas9 gene expression

注:M:100 bp marker;+:阳性小鼠;W:野生小鼠。图7 CDH1基因T7酶切活性鉴定结果Note.M: 100 bp marker.+: Positive mice.W: Wild mice.Figure.7 Results for identifying CDH1 gene T7 enzyme activity
CRISPR/Cas9技术是近几年快速发展的基因组编辑新技术,和锌指核酸酶 (zinc finger endonuclease, ZFN)、转录激活因子样效应物核酸酶(transcription activator-like effector nuclease, TALEN) 一样,都可以定向编辑细胞和个体的基因组序列。与其它两项技术相比,CRISPR/Cas9具有构建方便,高效,更多的可供靶向位点等优点[17-19]。此外,CAS9基因在特异性启动子的介导下,可以进行时空特异性的基因组编辑或基因调控,这为基因组织特异性编辑或调控提供了可能。传统组织特异性敲除主要依赖特异性表达CRE酶介导的基因片段删除,早期人们利用这种方法制备出多种乳腺特异性敲除小鼠模型[20],然而该技术需要进行多轮的杂交试验才能实现,尤其是多基因删除,需要更长的时间与大的实验群体才能得到。本实验应用CRISPR /Cas9 技术,以pX330质粒为骨架,将体外鉴定过能有效编辑BRCA1、BRCA2和CDH1三个基因的sgRNAs表达框同时连入一个表达载体,构建出可以同时编辑三个基因的共表达载体系统,并与乳腺组织特异性启动子-WAP启动子调控的CAS9表达框一起形成多基因组织特异性表达系统。因此,本研究利用该技术可一次性获得3个基因修饰的小鼠,避免了多轮杂交的过程,节省了时间和成本。
本次实验通过原核注射的方法获得了11只F0代的转基因阳性小鼠,其中3只为雌性小鼠。随后将F0代阳性鼠进行扩繁,采集部分泌乳期阳性小鼠不同组织样本,并检测CAS9基因表达与靶基因编辑情况,结果表明泌乳期乳腺组织能高表达CAS9基因,且CDH1基因产生了乳腺特异性剪切。而WAP启动子在泌乳期的其他组织也有微量表达,这与之前的研究类似[21]。本研究成功获得了乳腺组织特异性多基因编辑小鼠模型,与CRE酶介导BRCA1、BRCA2条件性敲除类似,该模型主要依赖于乳腺特异性启动子-WAP启动子的活性[22]。先前研究表明,在第一个妊娠期之前检测不到WAP启动子活性,而在妊娠中期(d15-d17)表达量激增[23-24],因此至少要经过一个妊娠泌乳期才能启动基因的表达[25-26]。尽管这会导致乳腺癌模型的制备周期延长,但这种体细胞累积性突变更加接近人类乳腺癌的发生、发展过程。我们知道绝大部分人类肿瘤的发生是靶组织中体细胞基因突变不断积累的结果,人们很难通过人体自身阐述肿瘤发生、发展的过程,这必须借助于实验动物。WAP启动子介导的CAS9基因表达,在特定时期乳腺的体细胞中不断累积BRCA1、BRCA2和CDH1基因突变,很好的模拟了乳腺癌的发生、发展的过程,为我们阐述乳腺癌的形成机理奠定了基础。此外,利用该方法制备的乳腺癌模型为药物靶点及靶向药物治疗提供了有利支持。
