新型Kdo2-lipid A合成菌株构建及其免疫生化活性研究
2018-08-20王碧雯李颜颜王小元
王碧雯 , 李颜颜 *,王小元
(1.食品科学与技术国家重点实验室,江南大学,江苏 无锡 214122;2.食品安全国际联合实验室,江南大学,江苏无锡 214122)
脂多糖 (Lipopolysaccharides,LPS), 俗称内毒素,是大多数革兰氏阴性细菌细胞外膜的主要组成部分[1-2]。LPS的结构分为2部分:疏水的Kdo2-lipid A 和亲水的多糖长链(Polysaccharides,PS),其在维持细胞外膜渗透性和稳定性上起到重要作用[3]。LPS能被哺乳动物细胞表面的Toll样受体4(TLR4)识别而激活先天性免疫系统、诱导促炎性细胞因子的释放。毒性高的LPS可引起促炎症细胞因子过量释放,引发热休克甚至死亡,而低毒性的LPS或其衍生物能有效激活先天性免疫系统但不会产生过量炎症分子,可作为免疫系统激活剂而被开发成疫苗或疫苗佐剂[4-5]。由于LPS提取工艺复杂、提取中需要用到大量的有机溶剂,如苯酚等,同时LPS分子量大、多糖长链部分结构各异,难以直接检测或定量,在生物免疫学研究及临床试验中有较大局限性。因此,降低LPS毒性、优化LPS结构、简化LPS提取检测步骤成为开发利用细菌内毒素的关键。
Kdo2-lipid A是LPS的生物活性中心和毒性中心,是细菌维持正常生长的最小LPS结构[5]。野生型大肠杆菌(Escherichia coli,E.coli)的 LPS 结构如图1所示,包含双磷酸、6条脂肪酸链的Kdo2-lipid A和一条多糖长链。Kdo2-lipid A与多糖长链的连接是由LPS合成途径中的waaC-waaF基因编码的2个庚糖转移酶WaaC和WaaF催化完成[6],前人研究利用四环素抗性基因插入waaC-waaF基因簇而使之失活,得到只合成短链LPS,即Kdo2-lipid A的菌株WBB06。Kdo2-lipid A相对分子质量小,具有双亲性,可通过三氯甲烷/甲醇体系简易萃取,并通过薄层层析的方法定量分析,在定性的质谱检测中更易电离、有高灵敏度,在水相体系中也有一定溶解度。因此,在疫苗或疫苗佐剂的开发应用中,Kdo2-lipid A较LPS更有优势。
本实验室前期构建了一株外源基因组成型表达菌HW003[7],该菌在E.coli W3110的染色体上敲除lacI基因,并lacZ位点中插入Kdo2-lipid A外源修饰基因lpxE(来自弗朗西斯菌株,编码的磷酸基团转移酶LpxE,能够特异性地去除Kdo2-lipid A的C1位磷酸基团[8])和pagL(来自沙门氏菌,编码的脱酰基酶PagL能够特异性地去除Kdo2-lipid A的C3棕榈酸脂肪酸链[9]),其合成的LPS包含特殊结构Kdo2-lipid A及多糖长链,较野生型W3110的LPS毒性降低。本研究在HW003菌株的基础上进一步精简LPS结构,敲除与糖链中庚糖合成相关waaC-waaF基因,构建了一株无抗性标记、能够直接合成不含多糖长链的、特殊结构Kdo2-lipid A的突变株BW003。随后研究了BW003菌株合成的Kdo2-lipid A的结构、菌株及其Kdo2-lipid A的细胞毒性及菌株相关生化表型,为进一步开发为疫苗佐剂及工业生产菌株奠定基础。
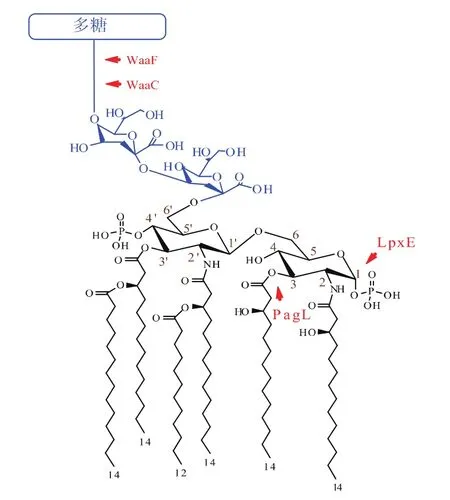
图1 E.coli LPS的结构及相关合成、修饰酶Fig.1 Structure of E.coli LPS and relative synthetic or modify enzymes
1 材料与方法
1.1 材料
1.1.1 实验菌株、质粒和引物 所涉及的实验大肠杆菌和辅助质粒列于表1,所涉及的引物列于表2。
1.1.2 培养基和培养条件 培养E.coli时均用LB培养基(蛋白胨 10 g/L,酵母膏 5 g/L,氯化钠 10 g/L,调pH到7.4);含pKD46或pCP20质粒的菌株在30℃下培养,筛选时需加入50 mg/L氨苄霉素;敲除片段同源重组转化子筛选时需加入30 mg/L卡那霉素;其它菌株均在37℃培养。
1.1.3 工具酶和主要试剂 PCR试剂盒,购买于广州东盛生物科技有限公司;Ex Taq聚合酶,购买于TaKaRa公司。
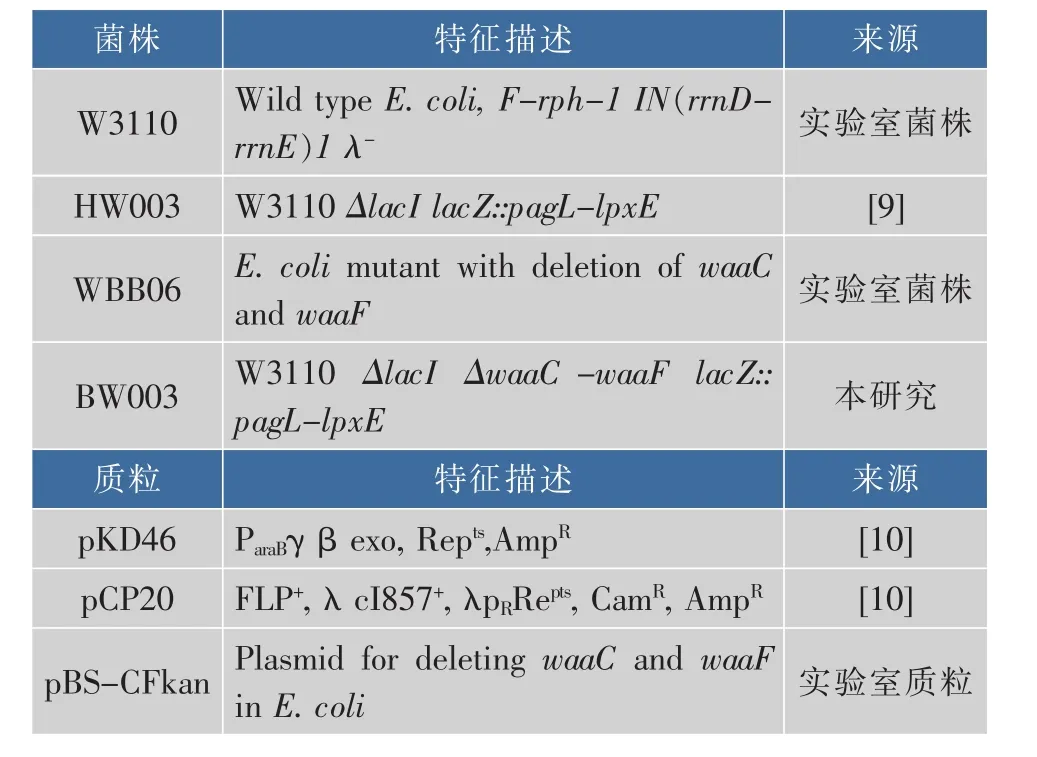
表1 本研究所使用的菌株和质粒Table 1 Strains and plasmids in this research

表2 本研究用到的引物Table 2 Primers in this research
核酸染料:GoldView,购买于上海赛百盛基因技术有限公司;核酸电泳 Marker,购买于Fermentas公司。DNA水解酶 I、RNA水解酶A和蛋白水解酶K,购买于上海生工生物工程有限公司。酵母膏干粉和蛋白胨,购买于英国OXIOID公司;Ampicillin和Kanamycin抗生素、L-阿拉伯糖、琼脂糖、琼脂粉,均购买于上海捷瑞生物有限公司;硅胶60薄层层析板,购买于德国默克公司;分析纯级别的氯化钠、三氯甲烷、甲醇等常规试剂,购买于国药集团。DMEM高糖培养基、RPMI-1640培养基、2.5 g/L胰酶、青霉素-链霉素溶液,购买于GIBCO公司;胎牛血清,购买于 Thermo-Hyclone公司;PMA、DMSO, 购买于Sigma-Aldrich公司;细胞培养瓶、细胞培养板,购买于 Corning公司;HEK-Blue hTLR4细胞、Normocin多效抗生素、HEK-Blue筛选液及HEK-Blue检测液,购买于Invivogen公司;THP-1细胞,购买于中国科学院细胞库;所有细胞因子ELISA试剂盒,购买于R&D公司。
1.2 方法
1.2.1 BW003菌株的构建 基于同源重组和位点特异性重组原理,waaC-waaF基因的敲除采用Datsenko[10]和Cherepanov等[11]报道的方法,敲除过程如图2所示。首先,在E.coli JM109中构建waaC-waaF基因的敲除质粒pBS-CFkan,再由PCR从敲除质粒上将敲除片段扩增出来、纯化、回收。在已构建好lpxE-pagL组成型表达的菌株HW003(W3110△lacI lacZ::lpxE-pagL)中转入pKD46辅助质粒。HW003/pKD46菌株在L-阿拉伯糖诱导培养、制备成感受态后将敲除片段转入,使其发生同源重组从而敲除目的基因。筛选出正确转化子后,通过高温42℃培养去除pKD46质粒,再在转化子中转入pCP20质粒促使位于kan片段两端的FRT位点发生位点特异性重组,从而去除kan片段。最后通过高温42℃培养去除pCP20质粒,进而得到无抗突变株BW003。E.coli感受态细胞制备方法和转化方法参照Han等[12]报道的方法。
1.2.2 Kdo2-lipid A与LPS的提取与纯化制备Kdo2-lipid A粗样采用Bligh-Dyer萃取法[13]。200 mL过夜培养菌液离心以收集菌体,所得菌体双蒸水洗涤一次后用38 mL三氯甲烷/甲醇/水一相体系(1∶2∶0.8,体积比)重悬菌体,磁力搅拌破碎1 h,离心分相,弃下相固体碎片、取上清液再加入10 mL三氯甲烷和10 mL水,配成三氯甲烷/甲醇/水二相体系(2∶2∶1.8,体积比);再次离心以分相,将下相液体移至旋蒸瓶,旋转蒸干溶剂,所得粗样-20℃保存。Kdo2-lipid A样品的纯化制备参照Zhou等[14]的DEAE-纤维素阴离子交换层析方法:将BW003的Kdo2-lipid A 粗样溶于 5 mL 三氯甲烷/甲醇/水(2∶3∶1,体积比)溶液,上样于一个事先用6倍体积三氯甲烷/甲醇/水 (2∶3∶1, 体积比) 溶液平衡的、10 mL DEAE-纤维素阴离子交换柱;用三氯甲烷/甲醇/240 mmol/L 醋酸铵(2∶3∶1,体积比)溶液洗脱磷脂等杂质,再用三氯甲烷/甲醇/480 mmol/L 醋酸铵(2∶3∶1,体积比)溶液将Kdo2-lipid A进行洗脱并收集;收集洗脱液配成三氯甲烷/甲醇/水二相体系 (2∶2∶1.8,体积比);离心取下相,旋蒸除去溶剂,得Kdo2-lipid A纯品,-20℃保存。

图2 W3110,HW003,BW003菌株染色体基因型比对和waaC-waaF敲除示意图Fig.2 Diagrams of chromosomes genotype alignment of strains W3110,HW003,BW003 and the deletion of waaC-waaF
LPS样品的提取采用热酚法[13]。离心菌液收集约5 g湿菌体,用双蒸水洗涤菌体一次后加入18 mL水和22 mL体积分数90%苯酚,68℃震荡1 h;冷却后4℃离心分相,取上相透析24 h以去除苯酚;透析后的溶液经真空冷冻干燥得到LPS粗样。LPS粗样按照Karow等[15]方法,复溶后添加DNA水解酶I、RNA水解酶A和蛋白水解酶K处理去除核酸及蛋白质,真空冷冻干燥后再用三氯甲烷-甲醇混合溶液洗涤以去除磷脂,最后复溶于双蒸水中真空冷冻冻干,得LPS纯品,4℃保存。
1.2.3 Kdo2-lipid A的薄层层析(TLC)分析及电喷雾电离质谱(ESI/MS)分析 薄层层析(TLC)分析及电喷雾电离质谱(ESI/MS)分析均按照Wang等[16]的方法,Kdo2-lipid A 粗样溶于三氯甲烷/甲醇(2∶1,体积比),TLC分析时将样品点于硅胶60 TLC板上,在三氯甲烷/甲醇/冰醋酸/水(25∶15∶2∶1,体积比)的展层剂中层析,层析结束后吹干板上残留展层剂,用乙醇/硫酸(1∶9,体积比)溶液进行碳化,最后加热至180℃显色;ESI/MS分析时,进样于WATERS SYNAPT Q-TOF Mass Spectrometer质谱仪,在120 V锥孔电压、负离子扫描模式下检测m/z 1 000~2 500范围的离子,所得数据用MassLynx V4.1 software软件处理分析。
1.2.4 HEK-Blue hTLR4细胞毒性分析 HEKBlue hTLR4细胞是经转染改造的人肾上皮细胞,是在HEK293细胞中表达了人TLR4及NF-kB诱导分泌的胚胎碱性磷酸激酶报告基因,能特异性识别LPS或含有LPS的细菌、实时监测LPS诱导的TLR4免疫通路的强弱,通过显色反应检测出对应样品的TLR4的刺激能力和对细胞的毒性。HEKBlue hTLR4细胞培养参照Needham等[17]的方法和细胞使用说明书。细菌菌体收集后用无热源磷酸缓冲液(PBS)洗2次后梯度稀释,先加入96孔板,无菌PBS作为空白对照;HEK-Blue hTLR4细胞收集计数后用HEK-BlueTM检测液重悬再接种至96孔板;细菌和细胞混合培养12 h后检测OD650的吸光度,吸光度与TLR4信号通路激起的强度成正比。
1.2.5 THP-1细胞的毒性分析 细胞培养参照Li等[18]的方法。细胞收获计数后,按终质量浓度20 ng/mL添加PMA并将细胞悬液接种至96孔板,培养12 h后细胞分化贴壁,倒弃旧培养基,更换新鲜培养基,同时加入质量浓度分别为 100、10、1、0.1 ng/mL的Kdo2-lipid A或LPS纯化样品,以不添加样品的新鲜培养基作空白对照。细胞再培养24 h后离心取培养上清液,保存于 -80℃。用于ELISA试剂盒测定细胞培养上清液中细胞因子 TNF-α、IL-8和RANTES的刺激释放量。
1.2.6 菌株生化表型分析
1)菌株细胞表面疏水性的测定。参照Del Re等[19]的方法,用PBS洗涤菌体一次并重悬,控制菌悬液OD600=0.5;取3 mL菌悬液置于试管中,再加入1 mL二甲苯,剧烈震荡2 min后静置0.5 h,取下相液体测OD600。

2)菌株外膜渗透性的测定。菌株外膜渗透性测定采用 NPN(N-Phenyl naphthylamine)方法[20]。离心收集菌体,用PBS洗涤菌体一次并重悬,控制菌悬液OD600=0.5;取1.92 mL菌悬液与80 μL 1 mmol/L的NPN溶液混匀于石英比色皿中,测定激发波长350 nm、发射波长420 nm、缝宽5 mm条件下的荧光吸收值。外膜渗透性与荧光吸收强度成正比。
3)菌株自凝集能力的测定。参照Rahman等[21]的方法,离心过夜培养菌液以收集菌体,用PBS洗涤菌体一次并重悬,控制菌悬液OD600≈2.0(记为A0,保留三位有效数字)。取10 mL菌液转移至刻度管,22℃静置 24 h,测定菌液 OD600值(记为 Ai,保留三位有效数字)。
自凝集百分数 AAg%=[(A0-Ai)/A0]×100
4)菌株抗生素MIC值的测定。将新生霉素(Novobiocin)用LB液体培养基倍比稀释,使质量浓度 分 别 为 1 000、500、250、125、62.5、31.3、15.6、7.80、3.90 μg/mL,加入96孔板中,留一排新型LB培养基作生长对照;另将等体积OD600=0.01的稀释菌液加入96孔板中,盖上盖子,37℃静置培养12 h,用酶标仪测定菌液OD600。将OD600小于0.15的点对应的抗生素浓度定义为菌株对新生霉素的MIC值。
2 结果与讨论
2.1 BW003菌株的构建
首先,对出发菌株HW003进行PCR鉴定(图3(a))。以lacZ-F/R(lacZ位点中间位置设计的一对引物)为引物,W3110基因组lacZ位点PCR产物大小为 300 bp 左右(图3(a)的 1 泳道);HW003 基因组PCR 产物则为 1 996 bp(图3(a)的 2 泳道),电泳结果验证表明出发菌株HW003的lacZ位点中成功插入lpxE-pagL基因。随后,通过同源重组的方法开始敲除waaC-waaF基因,成功敲除后,PCR验证图如图3(b)所示。 图3(b)1 泳道是以 waaC-waaF-F/R为引物,对HW003菌株基因组PCR得到的产物,大小为2 495 bp,是waaC-waaF基因未敲除的大小。泳道图3(b)2泳道是敲除片段同源重组后转化子基因组PCR所得产物,大小为1 668 bp。图3(b)3泳道则是去除kan片段后,BW003菌株基因组PCR产物,大小为635 bp,表明waaC-waaF基因敲除成功。
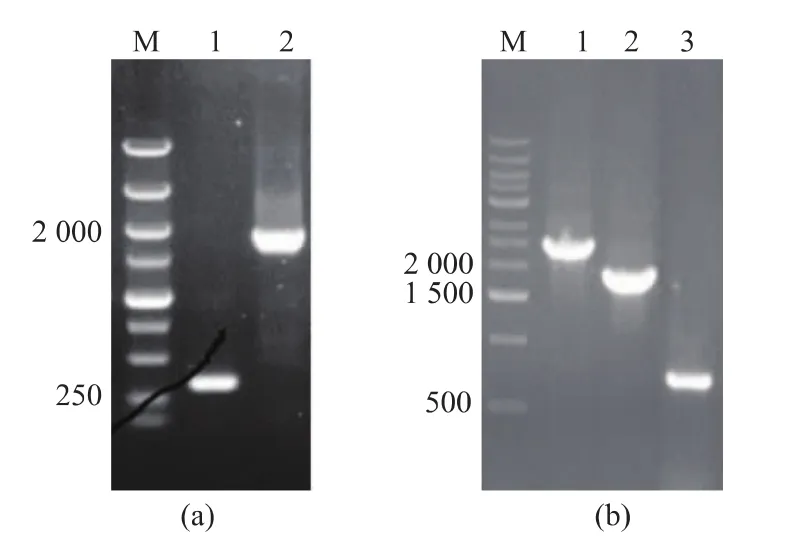
图3 各菌株lacZ位点和waaC-waaF基因段染色体PCR验证电泳图Fig.3 Verification of lazZ site and waaC-waaF gene by PCR
2.2 BW003菌株合成的Kdo2-lipid A结构鉴定
用已知 Kdo2-lipid A生产菌 WBB06(W3110 waaC-waaF::tet6)作为对照[6,22],根据 Bligh-Dyer 萃取法分别提取WBB06和BW003菌株合成的Kdo2-lipid A,样品分别点在硅胶60 TLC板上,层析显色,得图4(a)。根据展层剂性质,样品迁移速度与疏水性成正比,即磷酸基团越少、脂肪酸链越多迁移速度越快。磷脂相对分子质量小、疏水性最强,迁移速度最快,显色于TLC板上方。Kdo2-lipid A则集中于TLC板中部。WBB06粗样只含有单一的、双磷酸、6条脂肪酸链的Kdo2-lipid A,在TLC板上只有1个显色点;而BW003的粗样则呈现出上中下3个显色点,可见BW003合成了3种不同结构的Kdo2-lipid A。根据亲疏水性,推测中间一点为LpxE与PagL共同作用修饰后生成的目标产物-单磷酸、5条脂肪酸链的Kdo2-penteacyl-monophosphate-lipid A(Kdo2-P-MPLA);而目标产物下方的点与WBB06显色点平齐,则推测同为野生型双磷酸、6条脂肪酸链的Kdo2-lipid A;上方点则推测为LpxE作用,但PagL未作用而生成的单磷酸、6条脂肪酸链的Kdo2-monophosphate-lipid A(Kdo2-MPLA)。
通过电喷雾电离质谱(ESI-MS)对菌株BW003的粗样进一步进行结构鉴定。质谱结果与TLC分析相符合,BW003菌株粗样在质谱中检测出3个主要的[M-H]-负离子峰,分别是 m/z 1 930、2 156和 2 236,与平均相对分子质量为 1 931、2 157和2 237的Kdo2-P-MPLA、Kdo2-MPLA和 Kdo2-lipid A相对应。结果表明BW003菌株能够直接合成不含多糖长链的、特殊结构的Kdo2-P-MPLA,但同时混有部分Kdo2-lipid A和Kdo2-MPLA。
2.3 活菌刺激HEK-Blue hTLR4细胞的毒性分析
活菌直接刺激HEK-Blue hTLR4细胞后细胞液Abs650的吸光度正比于TLR4信号通路激起的强度。如图5所示,W3110,HW003及BW003活菌体都能有效激起TLR4信号通路;在菌浓度为103~106CFU/mL范围内,三者激起的TLR4信号强弱有明显差别:W3110最高,HW003适中,BW003最低;尤其在104 CFU/mL时,W3110的OD650高达1.02,HW003的OD650降低为0.66,而直接合成特殊结构Kdo2-lipid A的BW003菌株的OD650只有0.48,较W3110降低53%。研究结果表明BW003菌株LPS结构改变后,刺激细胞的毒性比野生型W3110菌株明显下降。

图4 BW003合成的Kdo2-lipid A薄层层析分析与电喷雾电离质谱分析Fig.4 TLC analysis and ESI/MS analysis of Kdo2-lipid A from BW003

图5 E.coli W3110,HW003及BW003活菌体对TLR4信号通路激起的强度比较Fig.5 Comparison of TLR4 stimulation of whole bacterial cells of E.coli W3110,HW003 and BW003
2.4 Kdo2-lipid A刺激THP-1细胞的毒性分析
为进一步研究LPS分子结构的改变对免疫细胞的毒性的影响,纯化后的W3110 LPS、HW003 LPS及BW003 Kdo2-lipid A样品被梯度稀释用于刺激人的THP-1巨噬细胞,并刺激后测定TLR4下游MyD88通路及TRIF通路的促炎症细胞因子TNF-α、IL-8和RANTES的释放量,细胞因子释放量越大说明细胞毒性越高。如图6所示,细胞因子释放量与样品质量浓度成正比,W3110 LPS诱导TNF-α、IL-8及RANTES的分泌量在4个浓度下最高,其毒性最强。而HW003 LPS与BW003 Kdo2-lipid A二者免疫活性接近,细胞毒性均较W3110有明显减弱。在样品质量浓度为100 ng/mL时,W3110 LPS诱导 TNF-α、IL-8及 RANTES的分泌量分别为 2 202,19 347,7 261 pg/mL; 而 BW003 则 只 有 1 563,14 175,5 621 pg/mL, 分别降低了 29%、27%和22%,说明BW003合成的特殊结构Kdo2-P-MPLA,虽然混合了其他2个结构,但较野生型的LPS能够明显减毒,同时也保持了一定的免疫活性,具有开发为新型疫苗佐剂的潜力。
2.5 菌株生化表型分析
以W3110和HW003作为对照,对BW003菌株的表型进行分析测定。由图7(a)可见,表达lpxE-pagL基因后HW003的疏水性较W3110(40%)略有提升,而表达lpxE-pagL基因并敲除waaC-waaF的BW003菌株由于其LPS同时缺失C1位磷酸基团、C3位脂肪酸链和多糖长链,菌株的外膜渗透性则明显升至W3110菌株的3倍,而对照的HW003菌株的外膜渗透性是W3110菌株的1.23倍。更高的外膜渗透性也使得抗生素更容易进入细菌细胞内部发生作用,因此BW003也表现出对抗生素的高度敏感,由图7(b)可见,W3110对新生霉素的MIC为1 000 μg/mL,HW003 略低,为 500 μg/mL,而BW003对新生霉素的MIC则骤降为15.6 μg/mL。另一方面,LPS结构的改变使得BW003菌株的细胞表面疏水性提升至83%(图7(c))。疏水性的增加使得菌株在液体培养体系中更容易聚团、凝集,在静置24小时后,BW003的自凝集比例为93%,而W3110和HW003仅为 23%和 22%(图7(d))。

图7 W3110、HW003及BW003菌株、外膜渗透性、新生霉素MIC、疏水性、自凝集能力的比较Fig.7 Comparison of membrane permeability,Novobiocin MIC,hydrophobicity and auto-aggregation of E.coli strains W3110,HW003 and BW003
3 结语
LPS能被哺乳动物细胞表面的TLR4受体识别激活先天性免疫反应,具有被进一步发展成为新型疫苗或疫苗佐剂的潜力。但LPS前期提取工艺复杂,消耗大量的苯酚,纯化步骤也相对繁琐,且由于LPS相对分子质量大、多糖长链部分结构各异,难以直接检测或定量,在生物免疫学研究及临床试验中有较大局限性。
本研究通过敲除HW003菌株中与多糖长链合成相关的waaC-waaF基因,使得多糖长链无法与Kdo2-lipid A相连接,成功构建一株直接合成新型、特殊结构的Kdo2-lipid A的突变株BW003。该菌株合成的LPS结构发生变化,其合成的新型Kdo2-lipid A可由三氯甲烷/甲醇/水体系简易快速萃取得到,不用消耗苯酚;其纯化步骤也较为便捷可行,通过DEAE离子交换柱梯度洗脱便可实现。Kdo2-lipid A可用TLC和ESI/MS直接分析、便于进行实时定量与检测,能够更好的应用于实验室或临床研究中。
BW003菌株合成的LPS结构定向改变后,菌株的外膜渗透性、抗生素敏感性、细胞表面疏水性及菌体自凝集能力较野生型W3110与HW003,均有一定程度的提高。外膜渗透性的改变加速了细胞内外物质的交换,菌体自凝集能力的增强简化了菌体收集的步骤,为后续在工业化发酵生产菌株的制备奠定了良好基础。
BW003菌株合成的Kdo2-lipid A具有多样性,共包括3种结构:Kdo2-P-MPLA (单磷酸、5条脂肪酸链)、Kdo2-MPLA(单磷酸、6条脂肪酸链)和Kdo2-lipid A(双磷酸、6条脂肪酸链);其合成产物的多样性是由LpxE和PagL 2个结构修饰酶作用效果不完全所致。新型特殊结构Kdo2-lipid A的存在,使得BW003菌体的毒性显著下降。HEK-Blue hTLR4细胞刺激实验结果表明BW003较野生型W3110的TLR4信号强度降低53%。纯化后BW003合成的Kdo2-lipid A分子刺激THP-1细胞后产生的炎症细胞因子TNF-α、IL-8和RANTES的浓度明显降低,表明Kdo2-lipid A分子较W3110的LPS而言,内毒素毒性减弱。研究结果为进一步探究不同结构内毒素的生物免疫活性、开发相关的新型疫苗及其疫苗佐剂奠定了基础。
