共表达分子伴侣PDI和Ero1对葡萄糖氧化酶在毕赤酵母中表达的影响
2018-08-18高庆华董聪王玥胡美荣王庆庆王云鹏罗同阳刘蕾
高庆华 董聪 王玥 胡美荣 王庆庆 王云鹏 罗同阳 刘蕾
(1. 河北省微生物研究所,保定 071051;2. 中国科学院微生物研究所,北京 100101)
葡萄糖氧化酶(Glucose oxidase,GOD)的系统命名为β-D-葡萄糖氧化还原酶(EC1.1.3.4),是一种需氧脱氢酶,专一催化β-D-葡萄糖生成葡萄糖酸和过氧化氢。GOD的催化特性使其具有去葡萄糖、脱氧、杀菌等功能,且安全无毒无副作用,是国家允许使用的酶制剂之一,在食品的加工保鲜、医学检测等方面都有广泛应用[1]。
GOD在自然界广泛分布,来源包括昆虫、红藻类、巧橘类水果、细菌和真菌等动植物、微生物体。目前用于生产的工业化菌种主要有黑曲霉和点青霉;但是对于工业化大规模生产来说,一个高效且经济的生产系统是迫切需要的[2]。毕赤酵母(Pichia pastoris)表达系统已经发展成为一个成熟的外源蛋白表达系统,已有上千种蛋白在毕赤酵母系统中得到成功表达。近些年,毕赤酵母被美国FDA认定为GRAS(Generally recognized as safe),为其在食品及医药上应用铺平了道路[3]。Crognale等[4]选择了甲醇营养型毕赤酵母作为宿主,将 GOD 基因整合入P. pastoris X33异源表达,通过3 L发酵罐培养发酵,GOD 活性到了 50 U/mL,相比野生型菌株产量提高了近 4倍。Guo等[5]将GOD基因整合到毕赤酵母中,摇瓶水平发酵酶活达到40 U/mL,是出发菌株A.niger Z-25的20倍。
随着毕赤酵母表达系统的广泛化应用,该系统的一些自身的缺陷也逐渐暴露出来。某些外源蛋白的表达水平过高,超过了宿主细胞翻译后处理加工能力所能承担的最大负荷,引起蛋白的错误折叠、不加工或错误定位,进而导致目的蛋白在内质网腔中形成多聚体,并阻滞在内质网妨碍其功能,或者分泌到胞外的蛋白质无活性。这些问题越来越成为阻碍毕赤酵母表达系统应用于高水平表达的瓶颈问题[2]。
研究表明,分子伴侣的大量表达可以促进蛋白折叠。内质网氧化还原酶(Endoplasmic reticulum oxidation 1,Ero1)和二硫键异构酶(Protein disulfide isomerase,PDI)均为内质网腔中的信号肽,其中PDI 的氧化活性帮助蛋白形成正确的折叠结构而分泌到胞外,对内质网腔内蛋白质二硫键的形成具有促进作用,帮助蛋白质分泌[6];Ero1在内质网中氧化还原态的PDI 成氧化态,间接帮助蛋白分泌[7]。陈飞等研究发现单独整合 PDI 及同时整合 Ero1、PDI 毕赤酵母菌株的 CiP酶活分别提高了 2.43 和2.62倍[8];陈凤祥等[9]在毕赤酵母中共表达Ero1和PDI分别使IFNβ-HSA 的表达量提高了80%和90%。
本研究中,在前期研究中我们已经克隆了一株来源于高产葡萄糖氧化酶点青霉的GOD基因,并筛选获得了毕赤酵母基因工程菌株X33-pMD-GOD[10],在此基础上,研究进一步通过共表达分子伴侣PDI基因及同时共表达分子伴侣Ero1-PDI基因对菌株的生长和分泌外源蛋白质GOD的影响。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 大肠克隆菌株DH5α购自天根生化科技有限公司;酵母表达菌株X33-pMD-AOXGOD由作者实验室构建并保藏;分子伴侣表达载体pPICZ A由中科院微生物研究所提供。
1.1.2 主要试剂和仪器 DNA聚合酶、T4 DNA连接酶和限制性内切酶BstB I、Not I、BamH I、Sfo I购自NEB公司;核酸 Marker购自上海生工;蛋白 Marker购自Thermo公司;G418和Zeocin购自Invitorgen 公司;其他试剂均为国产分析纯;质粒提取试剂盒、胶回收试剂盒均购自天根生化科技有限公司;PCR 仪购自东胜创新生物科技有限公司和Power B电泳仪和MINI P-4电泳系统购自凯元信瑞仪器有限公司公司;全自动凝胶成像仪购自上海天能科技有限公司;引物和DNA 测序由上海生工完成。
1.1.3 培养基 (1)毕赤酵母培养基:YPD和BMGY 参照文献配制。(2)分批发酵BSM培养基:参照文献配制。(3)补料培养基:含12 mL/L PTM1的50%甘油。(4)诱导培养基:含12 mL/L PTM1的100%甲醇,同时补加50%山梨醇。
1.2 方法
1.2.1 分子伴侣基因 PCR扩增 分子伴侣蛋白Ero1 和PDI 基因序列分别以GenBank公布的登录号XM_002489600.1和EU805807.1设计合成引物,引物两端均分别含有BstB I和Not I酶切位点(表1),以毕赤酵母的基因组为模板扩增出 Ero1和 PDI基因序列。PCR扩增条件:98℃ 5 min;98℃ 30 s,55℃30 s,72℃ 60 s,共 30 个循环 ;72℃ 10 min。
1.2.2 分子伴侣重组质粒构建 将PCR产物纯化后连接pGM-T克隆载体上,转化E.coli DH5α,提质粒进行测序鉴定。将得到的阳性克隆质粒采用BstB I和Not I双酶切,纯化后与同样双酶切载体pPICZ相连接,筛选阳性克隆验证,获得重组质粒pPICZ/Ero1和pPICZ/PDI。将基因测序正确的重组质粒pPICZ/PDI利用引物EP-F和EP-R扩增出含有AOX启动子、PDI基因以及终止子区域的目的片段,与测序正确且BamHI线性化的重组质粒pPICZ/Ero1进行同源重组,采用NOVO protein同源重组方法,构建克隆载体pPICZ/Ero1-PDI。转化E. coli DH5α感受态细胞,挑取单克隆进行PCR鉴定、测序,获得构建成功的重组质粒pPICZ/Ero1-PDI。

表1 分子伴侣基因PCR扩增引物列表
1.2.3 阳性转化子的获得 将测序正确的重组质粒pPICZ/PDI和pPICZ/Ero1-PDI分别用SfoI限制内切酶进行线性化后,分别电击转化同一株X33-pMDAOX-GOD感受态细胞,电转条件一样(1.5Kv、5 ms)。然后加入预冷的YPDS 30℃静置培养2-6 h后,涂布在终浓度 250 μg/mL G418和50 μg/mL Zeocin的YPD双抗平板上,置于30℃培养3 d后获得转化子。
1.2.4 重组蛋白的诱导表达 将重组菌株X33/pMDGOD和分别整合分子伴侣PDI,Ero1-PDI的X33/pMD-GOD菌株进行YPD平板划线培养,挑取单菌落接种于5 mL YPCS试管种子培养基中,20-24 h后加 1%(V/V)甲醇诱导,之后培养到48 h和 72 h各加1%(V/V)甲醇诱导,96 h收菌,8000 r/min离心 5 min收集上清,上清进行酶活检测。
1.2.5 毕赤酵母胞内蛋白的提取 收集酵母细胞(约1010个细胞酵母),以PBS缓冲液清洗两遍后,重悬于 450 μL毕赤酵母总蛋白提取缓冲液,加入等体积的酸处理过的玻璃珠,涡旋震荡 1 min,置于冰上 30 s,重复 10 次。4℃,16000×g,离心20 min,所得上清即为酵母胞内总蛋白提取液。采用BCA法定量蛋白上样量。
1.2.6 GOD的酶活测定 GOD 活性测定方法参照文献,采用邻一联(二)茴香胺分光光度法。1个酶活力单位是指在37℃、pH5.0条件下,在1 min内转化 1 μmol葡萄糖生成 1 μmol葡萄糖酸和 H2O2所需的酶量。
1.2.710 L发酵罐放大表达GOD 选取试管水平上葡萄糖氧化酶活性最高的转化子,在10 L发酵罐条件下进行GOD的表达。挑单克隆接种于 YPD培养基培养 10 h,按 3%接种量转接于 100 mL BMGY 培养基至OD600≈6接种于10 L发酵罐,初期装液量为 5.6 L BSM培养基,高压灭菌20 min后,以 10%发酵体积接种。期间每隔24 h 加维生素C 水溶液(终浓度80 μg/L),30℃培养,每12 h取样测定发酵液酶活;上清液进行10% 聚丙烯酰胺凝胶电泳,观察GOD 蛋白质的表达。具体方法参照文献[10]。
2 结果
2.1 共表达PDI、Ero1-PDI酵母菌株的构建
以P. pastoris X33基因组为模板,PCR扩增获得pdi和ero1基因,PCR产物经1%核酸电泳验证如图1-A,在1500 bp左右可见明显的特异性条带,其大小与目标序列预期相符。将纯化后的基因与克隆载体pMD 18-T连接后测序,测序结果与GenBank中收录的序列一致(GenBank编号:AJ302014和8197528)。
将测序正确的pMD 18-T-pdi、pMD 18-T-ero1和表达载体pICZ A同时进行BstB I和Not I双酶切,胶回收纯化后用T4连接酶16℃连接过夜,T1扩增,提取质粒后双酶切,酶切结果经1%核酸电泳分析,如图1-B所示,在2900 bp和1500 bp左右有特异性DNA条带,表明重组质粒构建正确,获得了克隆载体pMD-ERO1和pMD-PDI。
将基因测序正确的重组质粒pMD-PDI利用引物EP-F和EP-R扩增出含有AOX启动子、PDI基因以及终止子区域的目的片段,与测序正确且BamH I线性化的重组质粒pMD-ERO1进行同源重组,采用NOVO protein同源重组方法,构建克隆载体pMDERO1-PDI。
2.2 阳性转化子的筛选-试管水平检测分子伴侣对GOD表达的影响

图1 PCR扩增产物和重组质粒的鉴定
将X33/pMD-AOX-GOD菌株作为空白菌株,与分别整合分子伴侣PDI和Ero1-PDI的X33/pMDAOX-GOD菌株按照上述条件进行摇瓶发酵。空白菌株酶活14.376 U/mL,其中整合PDI的GOD菌株P-6和P-7酶活分别为22.539 U/mL和22.204 U/mL,酶活分别提高了56.8%和54.5%;同时整合Erol,PDI的GOD菌株EP-1、EP-8和EP-15酶活分别为达到21.001 U/mL、24.635 U/mL 和 25.321 U/mL( 图 2),酶活分别提高了46.0%、71.4%和76.1%。
由图2可知,整合PDI菌株少部分菌株的酶活还有降低的现象;整合Erol-PDI菌株蛋白表达均较对照有较大的提高。
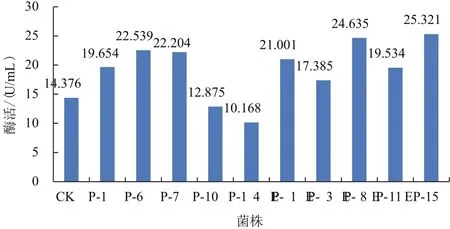
图2 共表达分子伴侣对GOD酶活的影响
进一步验证分子伴侣的表达量的多少是否与GOD的表达相关,研究了P-1、P-7、EP-8和EP-15共表达的分子伴侣的胞内表达和GOD表达的关系。从图3-A和图3-B中可以看出整合分子伴侣PDI和Ero1-PDI的X33/pMD-AOX-GOD菌株的PDI表达量明显高于对照菌株,菌株EP-15的PDI表达量最高,其目的蛋白GOD的表达量也最高,酶活也最高。

图3 葡萄糖氧化酶胞外表达(A)和分子伴侣胞内表达(B)检测
2.310 L发酵罐表达GOD
将对照菌株X33 /pMD-GOD和整合伴侣蛋白PDI及PDI/ERO1的菌株P-7和EP-15采用甲醇/山梨醇混合碳源流加方式进行10 L发酵罐放大培养。诱导表达144 h后下罐,发酵液上清进行SDS-PAGE分析(图4);对照菌株诱导156 h下罐时酶活为367 U/mL(图5-A),整合了分子伴侣的菌株蛋白表达量明显高于对照菌株(图5-B和C),同时整合伴侣蛋白Erol-PDI的EP-15菌株下罐酶活最高,诱导132 h 时酶活还未达到峰值,在156 h时达到了736 U/mL(图5-C),比对照菌株下罐时酶活提高了1倍。
3 讨论
毕赤酵母表达系统已经是一种成熟的外源蛋白表达系统,提高酵母表达外源蛋白水平的工艺方法研究一直备受关注[11-12]。通过在毕赤酵母X33 中分别单独表达分子伴侣PDI及同时共表达分子伴侣Ero1-PDI,探索了过量表达内质网中这两种蛋白质折叠辅助因子对外源蛋白质GOD分泌表达的影响。GOD由两个相同编码序列编码的同型二聚体分子构成,通过二硫键结合,二级结构中主要是β折叠[13]。已有研究表明,酵母细胞表达外源蛋白分泌产率与PDI 及二硫键形成有很大的相关性。酵母中 PDI 的过量表达能大幅度的提高外源蛋白的表达水平,尤其是含较多二硫键的蛋白[8,14]。另外,在毕赤酵母中表达Ero1使目的蛋白的表达量提高[9]。

图4 SDS-PAGE分析10 L发酵罐对照菌株(CK)和添加分子伴侣菌株GOD表达情况(P-7、EP-8及EP-15)
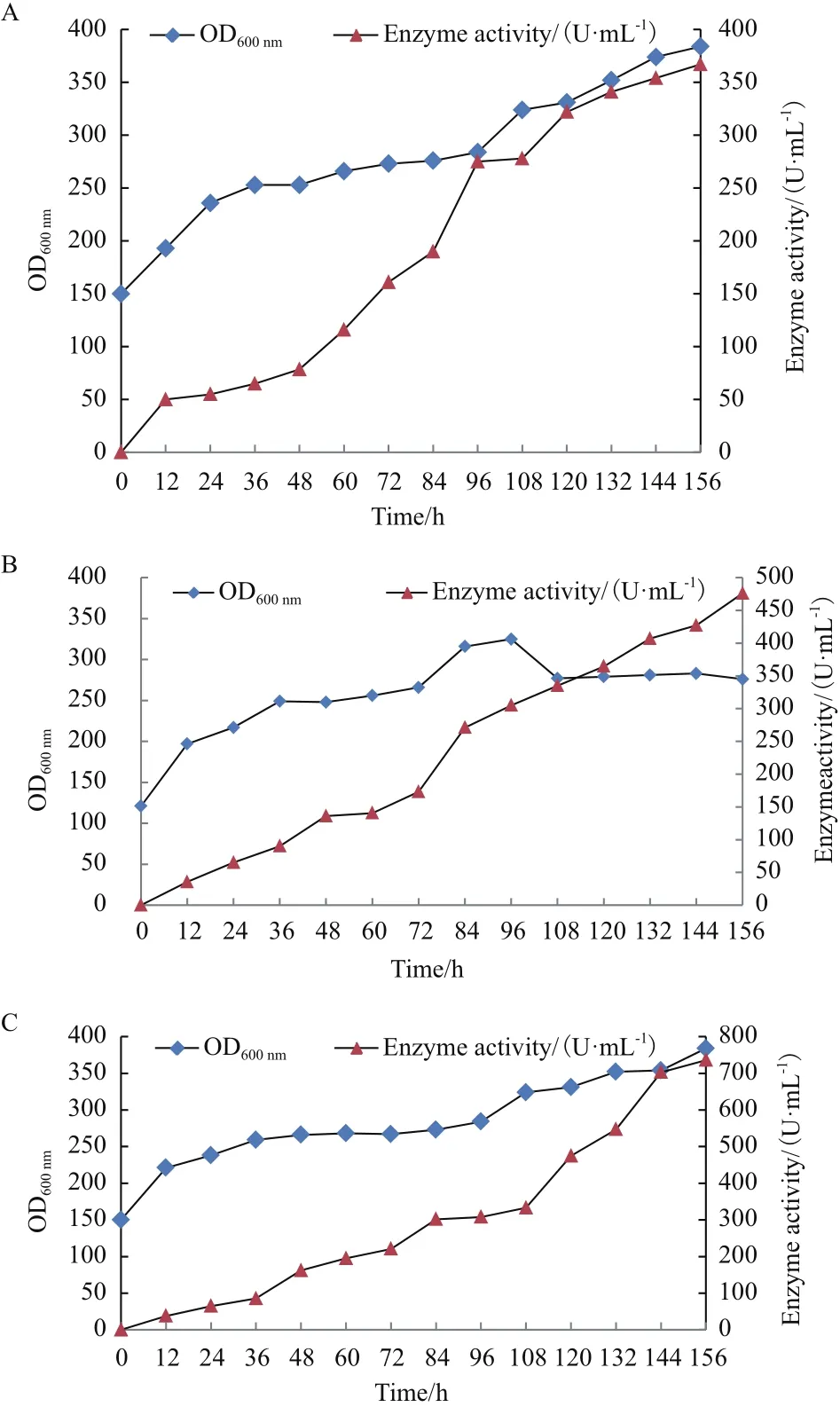
图5 GOD空白对照菌株((A)、添加分子伴侣菌株PDI及同时整合PDI-ERO1菌株(B)在10 L发酵罐中诱导生长和酶活分析
我们通过Ero1、PDI成功地在胞内过量表达,发现其首先并不影响宿主细胞的正常生长;其次在10 L 发酵罐放大实验研究发现单独整合PDI和同时整合Ero1-PDI 菌株酶活与表达量均有明显提高,其中整合Ero1-PDI 菌株的发酵酶活为736 U/mL 比对照菌株的发酵酶活367 U/mL整整提高了1倍。这说明当Ero1、PDI 以及GOD 共表达时,PDI 在帮助GOD形成二硫键时,本身被还原,Ero1 可以将PDI氧化,使其重新恢复功能,从而促进GOD 的表达。而且,分子伴侣的表达越多对其目的蛋白的表达越有利,这或与分子伴侣的表达也受甲醇诱导有关。但是,不是所有酶的表达,都与PDI的表达成正比[15]。
本研究通过整合分子伴侣促进蛋白正确折叠从而提高蛋白表达为以后在毕赤酵母细胞中的表达外源基因提供了新的思路和借鉴。分子伴侣是一个相互影响、相互作用的有机整体,如Ero1与PDI之间存在的相互作用,应该根据目的蛋白的结构特征,选择合适的分子伴侣优化表达组合。除此之外,还应该考虑分子伴侣与启动子、信号肽的组合及发酵工艺条件等,进行条件优化设计研究。
4 结论
本研究在工程菌X33/pMD-GOD中共表达分子伴侣PDI-Ero1 得到菌株X33/pMD-GOD/pPICZ-PDIEro1。在10 L 发酵罐放大实验过程中,采用甲醇/山梨醇混合碳源诱导的策略,GOD 最终酶活为736 U/mL,比对照菌株提高了1倍。
