四氢异喹啉阿魏酸衍生物的设计、合成及体外抗肿瘤活性研究
2018-08-02李家明张恩立陶兆林
王 杰,张 晖,李家明,张恩立,陶兆林
1.蚌埠医学院公共基础学院,蚌埠,233030;2.安徽中医药大学药学院,合肥,230031
近年来,随着分子技术和分离技术的不断发展,中药小分子的结构逐渐被认知,且一些小分子显示出较好的抗肿瘤活性。以天然小分子化合物为先导化合物,对其进行结构修饰和改造,开发高效、低毒的抗肿瘤药物受到了医药行业的高度关注。
四氢异喹啉天然小分子化合物是一种广泛存在于罂粟科、毛茛科等植物中的生物碱,近代药理学研究表明其具有抗肿瘤、抗病毒、保护神经、抗凝血等生物活性,其中抗肿瘤活性已成为人们研发的重点领域之一[1-3]。研究发现,诺斯卡品(结构如图 1)可通过抑制细胞分裂过程中的微管蛋白表达,发挥抗肿瘤作用,且毒副作用较小。郑剑斌等对其结构进行进一步修饰,合成了8个诺斯卡品衍生物,其中化合物1(结构如图 1)对人白血病细胞株HL-60显示了较好的抑制活性,IC50值为19.18 μM[4]。另有学者研究表明,磺胺类四氢异喹啉和酰胺类四氢异喹啉(结构如图 1)对人肝癌细胞株(HepG2)、人肺癌细胞株(A549)、人乳腺癌细胞株(MCF-7)和人结肠癌细胞株(HT-29)等癌细胞株具有较好的抑制作用[5-7]。综上所述,四氢异喹啉骨架可作为一种抗肿瘤先导物,对开发新型高效、低毒抗肿瘤药物具有重要的指导意义。
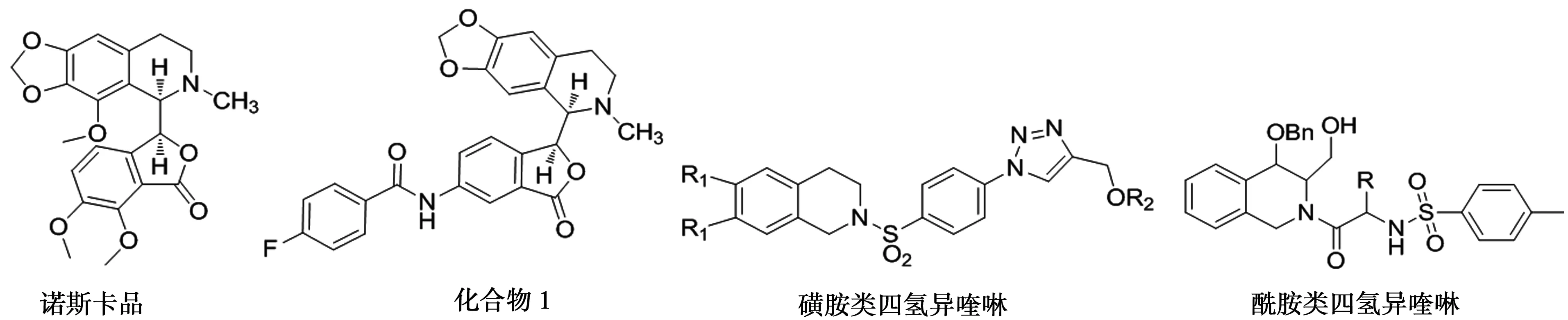
图1 诺斯卡品、化合物1、磺胺类四氢异喹啉和酰胺类四氢异喹啉的结构
苯丙烯酸类化合物主要包括阿魏酸及其衍生物、桂皮酸及其衍生物、咖啡酸及其衍生物等,此类化合物显示出较好的抗肿瘤活性,诱导癌细胞凋亡、抗血管新生及和细胞毒性为其主要作用机制[8-10]。
基于以上理论,同时依据药物化学中的分子片断理论和拼合原理,将四氢异喹啉骨架和阿魏酸进行拼合,设计了4个全新四氢异喹啉阿魏酸衍生物,以2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙胺、3,4,5-三甲氧基苯甲酸为起始原料,通过酰胺化反应、Bischler-Napieralski 反应和亲核取代反应对其进行合成,并通过SRB法对所合成化合物进行抗肿瘤活性初步筛选。合成路线如图2所示。

图2 四氢异喹啉阿魏酸衍生物的合成路线
1 实验部分
1.1 仪器、试剂与细胞株
H40数字熔点仪(国产)购自天津市精拓仪器科技有限公司;LCQ ADVANTAGE MAX 液质连用质谱仪购自FINNIGA 公司;IR-960傅里叶变换红外光谱仪(国产)购自天津瑞岸科技有限公司;CO2培养箱购自美国SHELLAB公司;Spectramax M2e酶标仪购自Molecular Devices公司;Bruker 400 MHz超导核磁共振仪(CDCl3为溶剂,TMS为内标) 。
3,4,5-三甲氧基苯甲酸纯度为99%、2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙胺纯度为97%、阿魏酸纯度为99%,均由合肥宝添科贸有限公司提供;乙醇、甲醇、二氯甲烷等常规试剂均分析纯购自合肥宝添科贸有限公司;秋水仙碱纯度为97%由合肥医工医药有限公司提供。
人脐静脉内皮癌细胞株(HUVEC)、MCF-7、HT-29由合肥医工医药有限公司提供。
1.2 合成方法
1.2.1 化合物6(5-(4-甲氧基苯基)-5,6,7,8-四氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉)的制备
在一250 mL圆底烧瓶中依次加入无水甲苯60 mL 和3,4,5-三甲氧基苯甲酸(3 g,14.2 mmol),常温搅拌澄清后加入SOCl215 mL,升温至75 ℃继续反应4 h,TLC[V(CHCl3)∶V(CH3OH)=10∶1] 检测反应完全,减压浓缩,用无水甲苯重蒸3次(30 mL/次),得化合物2,用15 mL CH2Cl2溶解备用。
在一250 mL三颈烧瓶中依次加入CH2Cl250 mL、化合物3(2.3 g,13.9 mmol)和三乙胺4 mL,冰浴条件下,将用CH2Cl2溶解的化合物2通过恒压滴液管缓慢加入。滴加完毕后,调为常温,然后继续反应2.5 h,TLC[V(乙酸乙酯)∶V(石油醚)=2∶1] 检测反应完全,用3 mol/L的HCl洗涤3次(50 mL/次),然后用1 mol/L的NaOH洗涤3次(80 mL/次),最后用饱和氯化钠溶液洗涤3次(60 mL/次),无水硫酸钠干燥,减压浓缩,用无水乙醇重结晶,抽滤,干燥,得白色固体化合物4(3.9 g),收率78.0%。
在一250 mL圆底烧瓶中依次加入无水甲苯40 mL 和化合物4(3.0 g,8.4 mmol),85 ℃搅拌下加入POCl3(6.2 mL,20 mmol),升温至115 ℃反应7 h,TLC[V(乙酸乙酯)∶V(石油醚)=2∶1]检测反应完全,浓缩,然后加入乙酸乙酯40 mL,转至分液漏斗,加水100 mL萃取,取水层,用浓氨水调至pH值为9-10,转移至分液漏斗,用CH2Cl2提取2次(40 mL/次),合并CH2Cl2层,用饱和食盐水洗涤3次(50 mL/次),无水硫酸钠干燥,浓缩,得淡黄色油状物5(2.3 g),收率82.1%,直接用于下步反应。
用无水甲醇20 mL将化合物5(2.3 g,6.8 mmol)溶解,冰浴条件下,将NaBH4(0.6 g,29 mmol) 分批3批缓慢加入,常温继续搅拌反应4 h,TLC[V(乙酸乙酯):V(石油醚)=2:1]检测反应完全,浓缩,用CH2Cl220 mL溶解,转移至分液漏斗,用冰水洗涤2次(50 mL/次),CH2Cl2层用无水硫酸钠干燥,浓缩,用甲醇重结晶,抽滤,干燥,得白色固体6(1.7 g),收率73.9%。
1.2.2 化合物9((E)-3-(4-乙酰氧基-3-甲氧基苯基)丙烯酰氯)的制备
在一150 mL烧杯中依次加入醋酸酐10 mL和浓硫酸2滴,常温搅拌15 min。一次性加入化合物7(阿魏酸)(10 g,50 mmol),搅拌10 min后变粘稠状,继续用玻璃棒搅拌半小时,FeCl3溶液检测显示反应基本完全,加水50 mL,搅拌,抽滤,将滤饼转移至150 mL烧杯中,加水40 mL,再用饱和碳酸钠溶液调pH值为9-10,抽滤。取滤液,滤液用3 mol/L的盐酸调pH值为2-3,析出白色固体,抽滤,用水洗涤3次(100 mL/次),干燥,得白色固体8(6.1 g),收率50.0%,m.p.192.4-194.1 ℃与文献值192.1-194.3 ℃基本一致[11]。
在一100 mL圆底烧瓶中依次加入无水甲苯30 mL、白色固体8(1.0 g,4.2 mmol)和DMF 2滴,常温搅拌澄清后加入SOCl210 mL,升温至75 °C继续反应4 h,TLC[V(CHCl3)∶V(CH3OH)=10∶1] 检测反应完全,减压浓缩,用无水甲苯带蒸3次(15 mL/次),得化合物9,用10 mL CH2Cl2溶解备用。
1.2.3 化合物11((E)-1-(5-(3,4,5-三甲氧基苯基)-7,8-二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)-3 -(4-羟基-3-甲氧基苯基)丙-2-烯-1-酮)的制备
在一250 mL三颈烧瓶中依次加入CH2Cl250 mL、化合物6 (1.4 g,4.0 mmol) 和三乙胺3 mL,冰浴条件下,将用CH2Cl2溶解的化合物9通过恒压滴液管缓慢加入,滴加完毕后,调为常温,然后继续反应2.5 h,TLC[V(乙酸乙酯)∶V(石油醚)=2∶1] 检测反应完全,用3 mol/L的HCl洗涤3次(30 mL/次),然后用1 mol/L的饱和碳酸钠溶液洗涤3次(40 mL/次),最后用饱和氯化钠溶液洗涤3次(40 mL/次),无水硫酸钠干燥,浓缩,得淡黄色固体10(1.5 g),收率68.2%。
在一100 mL圆底烧瓶中依次加入无水甲醇30 ml 和化合物10(1.2 g,2.1 mmol)和10 mL NaOH(24 mmol)甲醇溶液,室温过夜反应, TLC[V(乙酸乙酯):V(石油醚)=2:1]检测反应完全后,浓缩,用3mol/L的盐酸至pH值为4-5,抽滤,过硅胶柱,浓缩,干燥,得化合物11(0.61 g),收率61.0 %。
1.2.4 化合物12((E)-3-(4-(3-(二甲氨基)丙氧基)-3-甲氧基苯基)-1-(5-(3,4,5-三甲氧基苯基)-7,8 -二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)丙-2-烯-1-酮)的制备
在一100 mL圆底烧瓶中依次加入异丙醇15 mL KI (0.1 g,0.60 mmol)、K2CO3(0.2 g,1.5 mmol)和N,N-二甲基氯丙烷盐酸盐(0.2 g,1.3 mmol),常温搅拌反应2.5 h。同时在一100 mL三颈烧瓶依次加入异丙醇15 mL K2CO3(0.3 g,2.2 mmol)和化合物11(0.5 g,0.95 mmol),常温搅拌,将单颈烧瓶中的母液转移至三颈烧瓶,在80 ℃条件下继续反应4 h。TLC[V(氯仿)∶V(甲醇)=10∶1]检测反应完全,浓缩,过硅胶柱,浓缩,干燥,得黄色固体0.3 g,收率51.7%。
1.2.5 化合物13a((E)-3-(3-甲氧基-4-(3-吗啉丙氧基)苯基)-1-(5-(3,4,5-三甲氧基苯基)-7,8-二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)丙-2-烯-1-酮)的制备
在一100 mL三颈烧瓶中依次加入丁酮20 mL化合物11(0.5 g,0.95 mmol)、K2CO3(0.3 g,2.2 mmol),常温搅拌,通过恒压滴液漏斗缓慢加入丁酮1,3-二溴丙烷(0.2 mL,2 mmol)液,升温至85 ℃反应8 h,TLC[V(乙酸乙酯)∶V(石油醚)=2∶1]检测反应完全,抽滤,浓缩,得油状物0.5 g,将油状物用10 mL异丙醇溶解,转移至一100 mL三颈烧瓶中,然后依次加入吗啉 (0.2 mL,2.3 mmol)、K2CO3(0.2 g,1.5 mmol)、KI (0.1 g,0.60 mmol),升温至85 ℃反应4 h,TLC[V(乙酸乙酯)∶V(石油醚)=2∶1]检测反应完全,抽滤,浓缩,过硅胶柱,浓缩,干燥,得淡黄色固体13a(0.3 g),收率48.4%。
参照化合物13a合成方法得到目标化合物13b。
1.3 药理实验[12]
本药理实验选用的癌细胞株包括HT-29细胞、MCF-7细胞和HUVEC细胞,采用的方法为SRB法。具体步骤为:将3种对数生长期的癌细胞分别进行消化,吹打,制得单细胞悬液,在96孔培养板上分别进行接种,每孔接种5×103个细胞,加200 μL培养基;在培养箱中过夜培养(37 ℃、5%CO2);加入经过血浆孵育的梯度浓度的受试化合物,继续在培养箱中培养3 d;用10%三氯醋酸固定1 h;用双蒸水洗涤,干燥,然后在25 ℃条件下,每孔加入浓度为4 mg·mL-1的SRB溶液70 μL染色20分钟,使用1%醋酸洗涤,干燥;每孔加入浓度为10 mM 的Tris-Base溶液100 μL使SRB溶解;采用酶标仪测各孔OD值(检测波长:515 nm)。记录结果;计算抑制率:抑制率(%)=(OD对照-OD给药)/OD对照×100%,并计算IC50。
2 结果与讨论
2.1 目标化合物(11、12、13a、13b)的合成
所有目标化合物的结构均经1H-NMR、13C-NMR、MS、IR谱图佐证,目标化合物均为固体。在合成化合物5的过程中,滴加三氯氧磷的条件对反应具有较大影响,如在常温或温度高于95°C条件下加入,反应过程中产生黑色粘稠状物质,无化合物5生成,通过摸索,本文表述的条件为最佳。在化合物6的合成过程中,必须分批缓慢加入,因为此步为放热反应,且反应比较剧烈,如一次性加入过多,易导致溶液溅出。
化合物11:(E)-1-(5-(3,4,5-三甲氧基苯基)-7,8-二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)-3 -(4-羟基-3-甲氧基苯基)丙-2-烯-1-酮:白色固体, m.p.103.7-106.7 ℃。1H NMR (400 MHz,CDCl3) δ: 7.70(d,J=15.2 Hz,1H,CH=CH-Ar),7.11 (d,J=7.6 Hz,1H,ArH),6.99(s,1H,ArH),6.92(s,1H,ArH),6.90(d,J=6.8 Hz,1H,ArH),6.78(d,J=15.2 Hz,1H,CH=CH-Ar),6.58(s,1H,ArH),6.52(s,2H,CHN,ArH),5.94(d,J=15.2 Hz,2H,OCH2O),4.01(m,1H,NCH2CH2),3.91(s,3H,OCH3),3.82(s,3H,OCH3),3.77(s,6H,OCH3×2),3.57(m,1H,NCH2CH2),2.95(m,1H,NCH2CH2),2.80 (m,1H,NCH2CH2);13C-NMR (100 MHz,CDCl3) δ:153.1,153.0,147.6,146.8,146.3,146.2,143.5,138.2,137.5,137.4,128.2,127.7,127.6,121.9,114.9,114.7,110.1,108.6,108.2,106.3,100.9,60.8,56.3,56.0,55.7;IR (KBr,cm-1) υ:3431.0,2927.4,1640.1,1589.8,1510.5,1235.9,843.0,814.2; ESI-Mass forC29H29NO8: m/z (M-H)+518.18。
化合物12:(E)-3-(4-(3-(二甲氨基)丙氧基)-3-甲氧基苯基)-1-(5-(3,4,5-三甲氧基苯基)-7,8 -二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)丙-2-烯-1-酮:黄色固体,m.p.138.7-139.8°C。1H NMR (400 MHz,CDCl3) δ: 7.66(d,J=15.2Hz,1H,CH=CH-Ar),7.12(d,J=8.4Hz,1H,ArH),7.03(s,1H,ArH),6.88(d,J=8.8Hz,2H,ArH),6.82(d,J=15.2 Hz,1H,CH=CH-Ar),6.65(s,1H,ArH),6.57(s,1H,ArH),6.51-6.43(m,2H,ArH,CHN),5.95(d,J=15.2,2H,OCH2O),4.16(t,J=6.0Hz,2H,OCH2CH2),4.03-3.99(m,1H,NCH2CH2),3.89(s,3H,OCH3),3.82(s,3H,OCH3),3.77(s,6H,OCH3×2),3.57-3.52(m,1H,NCH2CH2),3.18(t,J=8.0Hz,2H,CH2CH2CH2N),2.96- 2.92(m,1H,NCH2CH2),2.81-2.77(m,1H,NCH2CH2),2.74(s,6H,CH3×2),2.37-2.31(m,2H,CH2CH2CH2);13C-NMR (100 MHz,CDCl3)δ:165.4,152.9,149.3,149.0,146.7,142.9,138.1,137.3,129.1,128.1,127.6,121.5,115.6,113.4,110.7,108.5,108.2,106.4,106.1,100.9,66.4,60.8,56.2,56.1,56.0,55.6,43.7,39.9,25.0;IR (KBr,cm-1) υ:3447.9,2935.7,2679.0,1638.1,1589.4,1508.3,1419.4,1234.5,1235.5,1124.4,1034.4,924.4,846.8; ESI-Mass forC34H40N2O8: m/z (M+H)+605.34。
化合物13a:(E)-3-(3-甲氧基-4-(3-吗啉丙氧基)苯基)-1-(5-(3,4,5-三甲氧基苯基)-7,8-二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)丙-2-烯-1-酮:淡黄色固体,m.p.155.4-157.3°C,1H NMR (400 MHz,CDCl3) δ: 7.68(d,J=15.2Hz,1H,CH=CH-Ar),7.10(d,J=8.4Hz,1H,ArH),7.03(s,1H,ArH),6.89-6.87(m,2H,ArH),6.77(d,J=15.2Hz,1H,CH=CH-Ar),6.64(s,1H,ArH),6.58(s,1H,ArH),6.51(s,1H,ArH),6.43(s,1H,CHN),5.94(d,J=15.6 Hz,2H,OCH2O),4.11(t,J=6.8Hz,2H,OCH2CH2),4.00-3.98(m,1H,NCH2CH2),3.89(s,3H,OCH3),3.82(s,3H,OCH3),3.77(s,6H,OCH3×2),3.72(t,J=4.4Hz,4H,NCH2CH2O),3.56-3.51(m,1H,NCH2CH2),2.95-2.88(m,1H,NCH2CH2),2.80-2.76(m,1H,NCH2CH2),2.53(t,J=7.2Hz,2H,CH2CH2CH2N),2.47(br,4H,NCH2CH2O),2.07-2.00(m,2H,CH2CH2CH2);13C-NMR (100 MHz,CDCl3) δ:165.5,152.9,150.0,149.4,146.2,143.2,138.1,137.3,128.3,124.1,121.7,115.0,112.6,110.5,108.6,108.1,106.1,100.9,67.1,66.7,60.8,56.2,56.0,55.5,55.3,53.5,39.9,29.4,26.0; IR (KBr,cm-1) υ:2935.7,1643.5,1590.3,1509.1,1235.2,1123.5,922.4,858.8;ESI-Mass forC36H42N2O9:m/z (M+H)+647.28。
化合物13b:(E)-3-(3-甲氧基-4-(3-哌嗪丙氧基)苯基)-1-(5-(3,4,5-三甲氧基苯基)-7,8-二氢-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-6(5H)-基)丙-2-烯-1-酮:13b黄色固体,收率39.2%,m.p.151.7-153.2°C。1H NMR (400 MHz,CDCl3) δ: 7.68(d,J=15.2 Hz,1H,CH=CH-Ar),7.10(d,J=8.0 Hz,1H,ArH),7.03(s,1H,ArH),6.89-6.87(m,2H,ArH),6.80-6.76(m,1H,ArH),6.64(s,1H,ArH),6.56(d,J=15.2 Hz,1H,CH=CH-Ar),6.51-6.43(m,2H,ArH,CHN),5.94(d,J=15.6 Hz,2H,OCH2O),4.10(t,J=6.8 Hz,2H,OCH2CH2),4.01-3.98(m,1H,NCH2CH2),3.89(s,3H,OCH3),3.82(s,3H,OCH3),3.77(s,6H,OCH3),3.56-3.51(m,1H,NCH2CH2),3.43-3.37(m,1H,NCH2CH2),2.81-2.72(m,1H,NCH2CH2),2.54-2.39 (m,10H,CH2CH2CH2N,NCH2CH2NH×2),2.02-2.01 (m,3H,CH2CH2CH2,NH);13C-NMR (100 MHz,CDCl3) δ:152.9,150.0,149.4,146.7,146.2,144.8,143.2,138.1,137.3,128.1,127.6,121.79,121.75,112.6,110.5,108.5,108.2,106.1,100.9,67.0,60.8,56.2,56.0,55.1,52.7,45.1,39.9,29.7,26.2; IR (KBr,cm-1) υ:3438.8,2932.8,2828.6,1641.5,1590.6,1508.9,1325.1,1235.3,1127.3,1034.5,924.6; ESI-Mass forC36H43N3O8: m/z (M+H)+646.28。
2.2 抗肿瘤活性
通过 SRB 法检测目标化合物体外抗肿瘤活性,结果表明,所合成的目标化合物对HT-29、MCF-7和HUVEC三种肿瘤细胞均具有不同程度的抑制作用,其中化合物12、13a、13b对HUVEC肿瘤细胞的抑制活性与秋水仙碱相当。通过目标化合物的结构和初步抑制活性筛选结果可看出,阿魏酸羟基通过醚键分别与哌嗪、吗啉、叔胺结合,对HUVEC的活性逐渐增强,且均高于裸露羟基。因此,我们推测,阿魏酸羟基上引入含氮侧链可以提高化合物的抗肿瘤活性,且脂肪胺侧链的化合物抗肿瘤活性优于含氮杂环的化合物,因此我们后期将对含脂肪胺侧链进行深入研究,以期获得较好活性的化合物。详见表1。


化合物IC50/μMHUVECMCF-7HT-291112.95±4.698.50±3.778.66±1.82121.71±0.392.44±0.441.77±0.7213a3.34±1.8712.43±5.662.45±0.8513b4.05±0.2912.88±6.952.84±1.59秋水仙碱1.88±0.431.04×10-2±0.28×10-23.22×10-2±1.30×10-2
3 结 语
本文以3,4,5-三甲氧基苯甲酸、2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙胺和阿魏酸为起始原料,通过酰化反应、Bischler-Napieralski 反应、还原反应、酯化反应、水解反应、亲核取代反应合成了4个四氢异喹啉阿魏酸衍生物,对所合成的4个目标产物的结构进行了1H-NMR、13C-NMR、MS、 IR谱图表征,均为所设计化合物,并测试了化合物其对HUVEC、MCF-7、HT-29的抑制活性,目标产物均显示了一定的抗肿瘤活性。
