高效液相色谱快速测定草本植物饮料中28种外源性药物和内源性成分
2018-08-02何嘉雯温家欣赖宇红曹雅静
何嘉雯, 温家欣, 赖宇红, 曹雅静
(广东省药品检验所, 广东 广州 510180)
草本植物饮料凭借着去火排毒的宣传,以功能性饮料的姿态成为饮料市场中的新宠,人们熟知的有凉茶、板蓝根饮料等[1]。随着草本植物饮料的兴起,凉茶作为重要代表再次在市场上火起来。然而在激烈的市场竞争中,不法商家在凉茶中非法添加化学药物,以求达到其宣称的降火功效,埋下了严重的安全隐患。这种违法行为并不是个别行为,已渐渐演变成行业潜规则,严重影响草本饮料产业健康发展,扰乱市场秩序。
目前,针对草本植物饮料非法添加的研究较少[2-9]。具有解热、消炎药理作用的药物均有被非法添加的可能,目前发现的药物有非甾体解热镇痛药、激素、抗菌药、中枢兴奋剂等[10-14],种类繁杂。谢文辉等[3]和谢集照等[4]采用TLC、刘辉等[5]采用酶联免疫法,建立了对乙酰氨基酚的快筛方法。温家欣等[6]采用HPLC快速测定凉茶中11种非法添加化学药物。宋宁宁等[7]建立了QuEChERS-UPLC-MS/MS测定凉茶中12种化学药物。韩婉清等[8]采用SPE-UPLC-MS/MS测定凉茶中15种植物源性兴奋剂和外源性药物。通过QuEChERS、SPE净化手段去除基质干扰,使液相色谱-质谱联用法获得较好的结果。然而,本研究组在大批量实际样品检测过程中发现,液相色谱-质谱联用法净化手段虽能减少基质污染,但凉茶中外源性药物过高的浓度造成的自身污染仍难以避免,使得仪器污染后出现大量假阳性,影响实验重复性。虽然稀释样品能改善污染情况,但由于不同药物间添加量的极大差异和非法添加的未知性,稀释容易造成漏检。而且目前研究集中在液体凉茶,没有涵盖颗粒剂、凉茶粉、代用茶等新剂型。本文以添加风险较高的28种成分作为研究对象,选择抗污染能力强、重复性好的高效液相色谱法,建立一套准确性好、耐用性高的检测方法,为草本植物饮料的监督检验提供助力。
1 实验部分
1.1 仪器与试剂
Waters e2695高效液相色谱仪,配二极管阵列检测器(美国Waters公司); Thermo Accucore C18色谱柱(100 mm×4.6 mm, 2.6 μm,美国Thermo公司); Sartorius MS205DU电子天平(德国Sartorius公司); Milli-Q超纯水系统(美国Millipore公司)。
标准品纯度均≥98% ,其中对乙酰氨基酚(paracetamol)、水杨酸(salicylic acid)、咖啡因(caffeine)、安乃近(dipyrone)、4-甲氨基安替比林(4-methylaminoantipyrine)、甲氧苄啶(trimethoprim)、氨基比林(antipyrine)、磺胺甲噁唑(sulfamethoxazole)、非那西丁(phenacetin)、泼尼松(prednisone)、氢化可的松(hydrocortisone)、吡罗昔康(piroxicam)、异丙安替比林(propyphenazon)、地塞米松(dexamethasone)、舒林酸(sulindac)、酮洛芬(ketoprofen)、萘普生(naproxen)、醋酸泼尼松(prednisone acetate)、芬布芬(fenbufen)、醋酸地塞米松(dexamethasone acetate)、奥沙普秦(oxaprozin)、萘丁美酮(nabumetone)、双氯芬酸钠(diclofenac sodium)、保泰松(phenylbutazone)、吲哚美辛(indometacin)、布洛芬(ibuprofen)和阿司匹林(aspirin)均购自中国食品药品检定研究院;甲基泼尼松龙(methylprednisolone)购自德国Dr. Ehrenstorfer公司。甲醇和乙腈(均为色谱纯)购自美国Holly公司,乙酸铵和冰醋酸(均为分析纯)购自广州化学试剂厂,实验用水为超纯水。
草本饮料样品收集于2016年~2017年风险监测任务,范围涵盖广东全省,包含预包装产品和现场制售凉茶,共456批次。
1.2 标准溶液的配制
标准储备液:除阿司匹林外,分别准确称取各成分标准品0.02 g,用甲醇溶解并配制2 g/L的标准储备液。吡罗昔康难溶,可用冰醋酸助溶。阿司匹林采用甲醇-冰醋酸(99∶1, v/v)配制。
混合标准溶液:分别吸取一定量的标准储备液,用甲醇-水(70∶30, v/v)稀释,混匀,配制成质量浓度为50 mg/L的混合标准溶液,现配现用。定量时,阿司匹林需用甲醇-冰醋酸(99∶1, v/v)单独配制。
混合标准工作溶液:吸取适量混合标准溶液,用甲醇-水(70∶30, v/v)配制成1~50 mg/L的系列混合标准溶液,现配现用。必要时,可按实际需要,采用分组方式配制质量浓度范围更广的混合标准工作溶液。
1.3 样品前处理
液体样品:摇匀,称取5.0 g试样,加甲醇-水(70∶30, v/v)适量。超声提取15 min,放冷,定容至25 mL,摇匀,提取液过0.22 μm滤膜,待测。固体饮料、代用茶和颗粒:样品先用研磨机充分粉碎或用研钵研细,称取0.5 g试样,加甲醇-水(70∶30, v/v)适量,超声提取30 min,放冷,定容至50 mL,摇匀,提取液过0.22 μm滤膜,待测。若样品中待测成分浓度超出线性范围,用甲醇-水(70∶30, v/v)适当稀释。
1.4 分析条件
色谱柱:Thermo Accucore C18色谱柱(100 mm×4.6 mm, 2.6 μm);流速为1.2 mL/min;柱温为35 ℃;进样量为5 μL;检测波长为254和220 nm。流动相:A为甲醇,B为乙腈,C为20 mmol/L乙酸铵溶液(pH 4.2,冰醋酸调)。梯度洗脱条件:0~3 min, 3%A~6%A, 6%B~12%B, 91%C~82%C; 3~4 min, 6%A~8%A, 12%B~15%B, 82%C~77%C; 4~9 min, 8%A~12%A, 15%B~23%B, 77%C~65%C; 9~12 min, 12%A, 23%B, 65%C; 12~24 min, 12%A~20%A, 23%B~38%B, 65%C~42%C; 24~25 min, 20%A~3%A, 38%B~6%B, 42%C~91%C; 25~30 min, 3%A, 6%B, 91%C。
1.5 计算公式
除阿司匹林外,27种成分计算公式为:
(1)
其中X为草本植物饮料中化学成分的含量(mg/kg),C为由标准曲线得出的质量浓度(mg/L),V为稀释体积(mL),m为试样取样量(g)。
阿司匹林在溶液中易水解,计算时需对降解产物水杨酸进行折算,计算公式为:
(2)
其中asp.表示阿司匹林,s.a.表示水杨酸。仅检出水杨酸时,视为阿司匹林未检出,不进行折算。
2 结果与讨论
2.1 色谱条件的优化
2.1.1色谱柱的选择
核壳填料能减少分析物在色谱柱中的涡流扩散,从而获得较好的分离效率,缩短分析时间,实现快速分离[15]。考察了不同品牌和不同填料的C18核壳色谱柱的分离效果,包括Thermo Accucore C18(100 mm×4.6 mm, 2.6 μm)、Thermo Accucore aQ(100 mm×4.6 mm, 2.6 μm)、Thermo Accucore RP-MS(100 mm×4.6 mm, 2.6 μm)、Agilent Poroshell 120 EC-C18(100 mm×4.6 mm, 2.7 μm)和ACE UltraCore SuperC18(100 mm×4.6 mm, 2.5 μm)。图1为分别使用5种色谱柱的分离情况比较。Thermo Accucore C18、Thermo Accucore aQ和Thermo Accucore RP-MS为同品牌的不同C18填料色谱柱,在相同的色谱条件下保留行为基本相似,Thermo Accucore C18能基本分离28种成分,但Thermo Accucore aQ在5~10 min、Thermo Accucore RP-MS在20~25 min分离效果不理想。Agilent Poroshell 120 EC-C18除氢化可的松和吡罗昔康分离较差、保泰松和吲哚美辛重合外,其余24种成分分离良好。ACE UltraCore SuperC18除萘丁美酮和双氯芬酸钠分离较差外,其余26种成分能基本分离。综上,选择Thermo Accucore C18作为分析柱。

图 1 使用不同色谱柱时28种成分的分离色谱图Fig. 1 Chromatograms of the 28 components separated on different columns a. Thermo Accucore C18 (100 mm×4.6 mm, 2.6 μm); b. Thermo Accucore aQ (100 mm×4.6 mm, 2.6 μm); c. Thermo Accucore RP-MS (100 mm×4.6 mm, 2.6 μm); d. Agilent Poroshell 120 EC-C18 (100 mm×4.6 mm, 2.7μm); e. ACE UltraCore SuperC18 (100 mm×4.6 mm, 2.5 μm). Peak Nos: 1. paracetamol; 2. salicylic acid; 3. caffeine; 4. aspirin; 5. dipyrone; 6. trimethoprim; 7. antipyrine; 8. 4-methylaminoantipyrine; 9. sulfamethoxazole; 10. phenacetin; 11. prednisone; 12. hydrocortisone; 13. piroxicam; 14. propyphenazon; 15. methylprednisolone; 16. dexamethasone; 17. sulindac; 18. ketoprofen; 19. naproxen; 20. prednisone acetate; 21. fenbufen; 22. dexamethasone acetate; 23. oxaprozin; 24. nabumetone; 25. diclofenac sodium; 26. phenylbutazone; 27. indometacin; 28. ibuprofen.
2.1.2缓冲盐pH值的优化
由于28种成分结构各异,缓冲盐pH值的变动对酸碱物质的保留行为有较大影响,可能改变出峰顺序。考察pH 4~pH 5之间各成分的保留行为。随着pH值降低,保泰松、吡罗昔康、阿司匹林、舒林酸、酮洛芬、萘普生、芬布芬、奥沙普秦、双氯芬酸钠、吲哚美辛、布洛芬的保留时间增加,氨基比林、4-甲氨基安替比林的保留时间减少,其余成分的保留时间变化不大。当缓冲盐pH值为4.2时,分离效果最佳。
2.1.3光谱特征和检测波长的选择
阿司匹林、水杨酸、布洛芬在220 nm下有较强吸收。但受梯度影响,在220 nm下的色谱基线波动较大,阿司匹林、水杨酸的积分效果较差。其他成分最大吸收波长分布在240~290 nm之间,在254 nm下均有良好的响应。综合考虑,选取254 nm作为主要检测波长,220 nm作为布洛芬的辅助检测波长。
2.2 前处理的优化
2.2.1提取溶剂的选择
28种成分均能溶于甲醇、乙醇、丙酮等有机溶剂,综合考虑液相色谱的适用性,提取溶剂考察以甲醇、乙腈为主。对于固体样品,考察了水、甲醇-水(70∶30, v/v)、甲醇、甲醇-乙腈(50∶50, v/v)的提取效果。部分成分在水中溶解度较差,回收率不理想;有机试剂提取效果佳,但提取液有较强的溶剂效应,峰形差。甲醇-水(70∶30, v/v)作提取溶剂时回收率达92%以上,同时避免高有机溶剂比例带来溶剂效应,令人满意。部分液体样品沉淀较多或过于浓稠,直接过滤干扰非常大。同法考察液体样品的提取溶剂,结果与固体样品类似,甲醇-水(70∶30, v/v)作提取溶剂时回收率达88%以上。因此选择甲醇-水(70∶30, v/v)作为提取溶剂。
2.2.2易降解成分的稳定性考察
阿司匹林、安乃近在溶液中稳定性较差,对其稳定性进行考察。阿司匹林的降解物主要为水杨酸,安乃近的降解物主要为4-甲氨基安替比林,其降解程度与溶剂和基质有关。在甲醇-水(70∶30, v/v)中,阿司匹林24 h内降解64% ,安乃近24 h内降解9% 。将空白样品按1.3节方法进行前处理,制得空白样品溶液,在该溶液中阿司匹林24 h内降解90% ,安乃近24 h内降解10% 。在甲醇-冰醋酸(99∶1, v/v)中,阿司匹林24 h内几乎不降解。因此,阿司匹林标准溶液采用甲醇-冰醋酸(99∶1, v/v)配制,现配现用。当计算阿司匹林含量时,将降解的水杨酸进行折算,更为合理。本实验标准溶液和样品溶液配制后应在24 h内完成分析。
2.3 方法学评价
2.3.1线性范围与检出限
除阿司匹林外,准确吸取适量混合标准溶液,用甲醇-水(70∶30, v/v)配制质量浓度为1、5、10、25、40和50 mg/L的系列溶液。阿司匹林采用甲醇-冰醋酸(99∶1, v/v)配制,质量浓度为5、10、25、40、50和100 mg/L。以峰面积(y)对质量浓度(x, mg/L)进行线性回归,结果表明各成分呈良好的线性关系,相关系数(r)均大于0.999(见表1)。
当浓度接近检出限时,应考虑样品基质的干扰,配制基质标准溶液以确定检出限(LOD)。将空白液体样品和固体饮料样品按1.3节方法进行前处理,制得溶液作为检出限溶液稀释溶剂,以3倍信噪比(S/N=3)的结果作为检出限。结果表明,液体样品和固体样品中成分的检出限分别为1~10 mg/kg和20~200 mg/kg(见表1)。
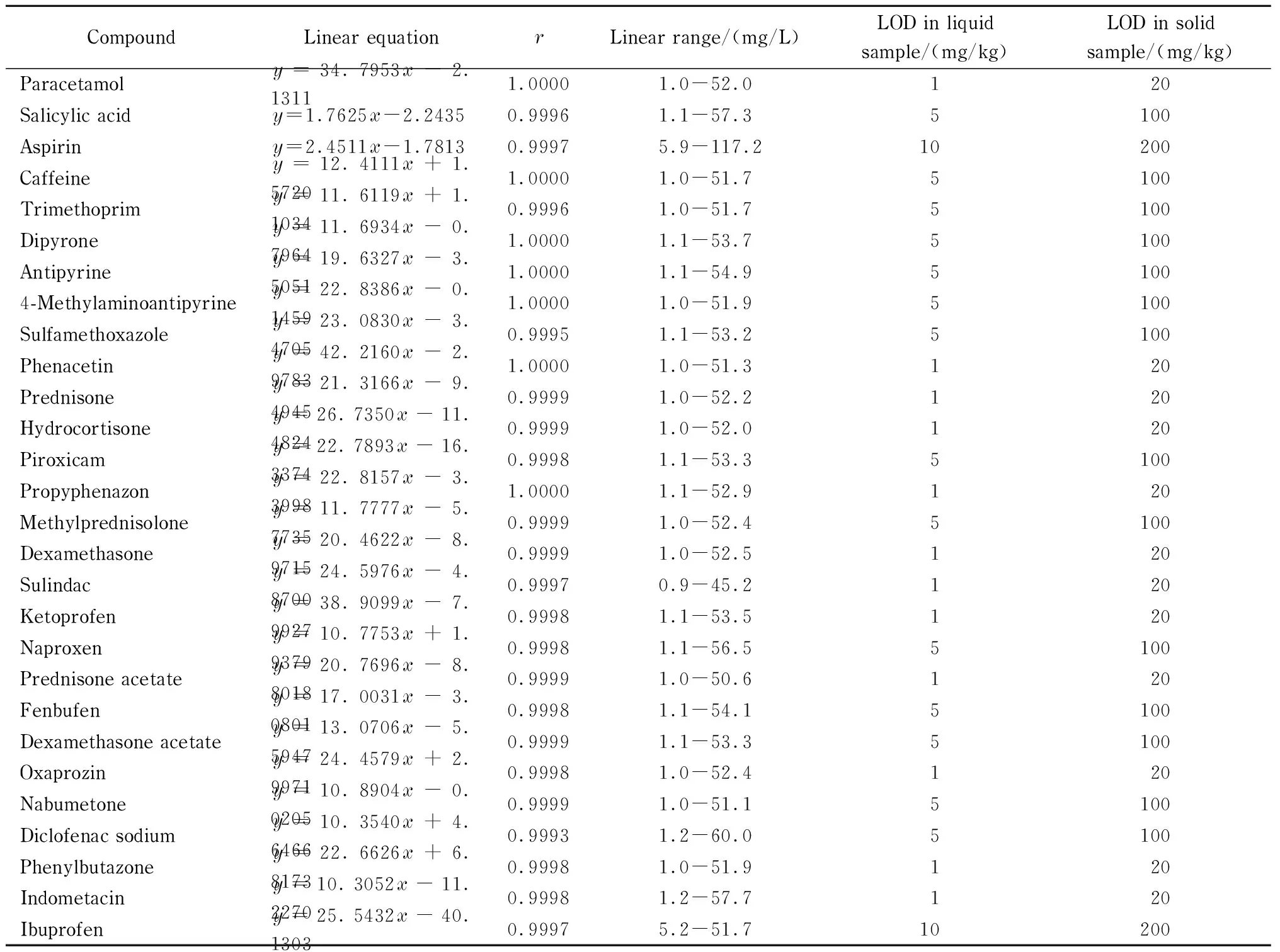
表 1 28种成分的线性方程、相关系数(r)、线性范围与检出限Table 1 Linear equations, correlation coefficients (r), linear ranges and limits of detection (LODs) of the 28 components
y: peak area;x: mass concentration, mg/L.
2.3.2回收率与精密度
按照1.3节样品前处理方法向空白基质中分别加入低、中、高3个水平的混合标准溶液进行加标回收率的测定。结果(见表2)表明,液体样品中28种成分的加标回收率为88.8% ~118.6% , RSD为0.1% ~6.7% ;固体样品中的平均加标回收率为92.7% ~112.3% , RSD为0.1% ~6.4% 。
2.4 实际样品检测
2.4.1检测结果
应用建立的方法对456批次样品进行分析,阳性样品代表色谱图见图2。结果表明,55批次样品中检出目标成分,检出率为12.1% 。检出成分包括对乙酰氨基酚、阿司匹林、咖啡因、非那西丁等。对乙酰氨基酚检出批次最多,共48批次。普遍存在组合添加的情况,具体结果见表3。

图 2 阳性样品色谱图Fig. 2 Chromatogram of the positive sample Peak Nos: 1. paracetamol; 9. sulfamethoxazole; 20. prednisone acetate.
2.4.2检测结果分析
统计实验结果可知,不同种类成分的添加量是不同的,可基本分为两级浓度水平。第一级是以对乙酰氨基酚为代表的解热镇痛药,添加范围为50~4 500 mg/kg(液体样品)和1.0×104~4.5×105mg/kg(固体样品),其特点是浓度较高、跨度较大、个别成分添加量极高。第二级为以醋酸泼尼松为代表的糖皮质激素,添加范围为8~20 mg/kg(液体样品)和6.0×102~6.0×103mg/kg(固体样品)。这种非法添加特点与药物治疗剂量相关。当两种浓度水平成分被同时添加于同一样品时,需选择合适的稀释倍数,避免峰过载或低含量成分漏检。另外,部分第一级成分也存在低浓度检出的情况,可能是非法添加现场加工器皿不洁造成的。
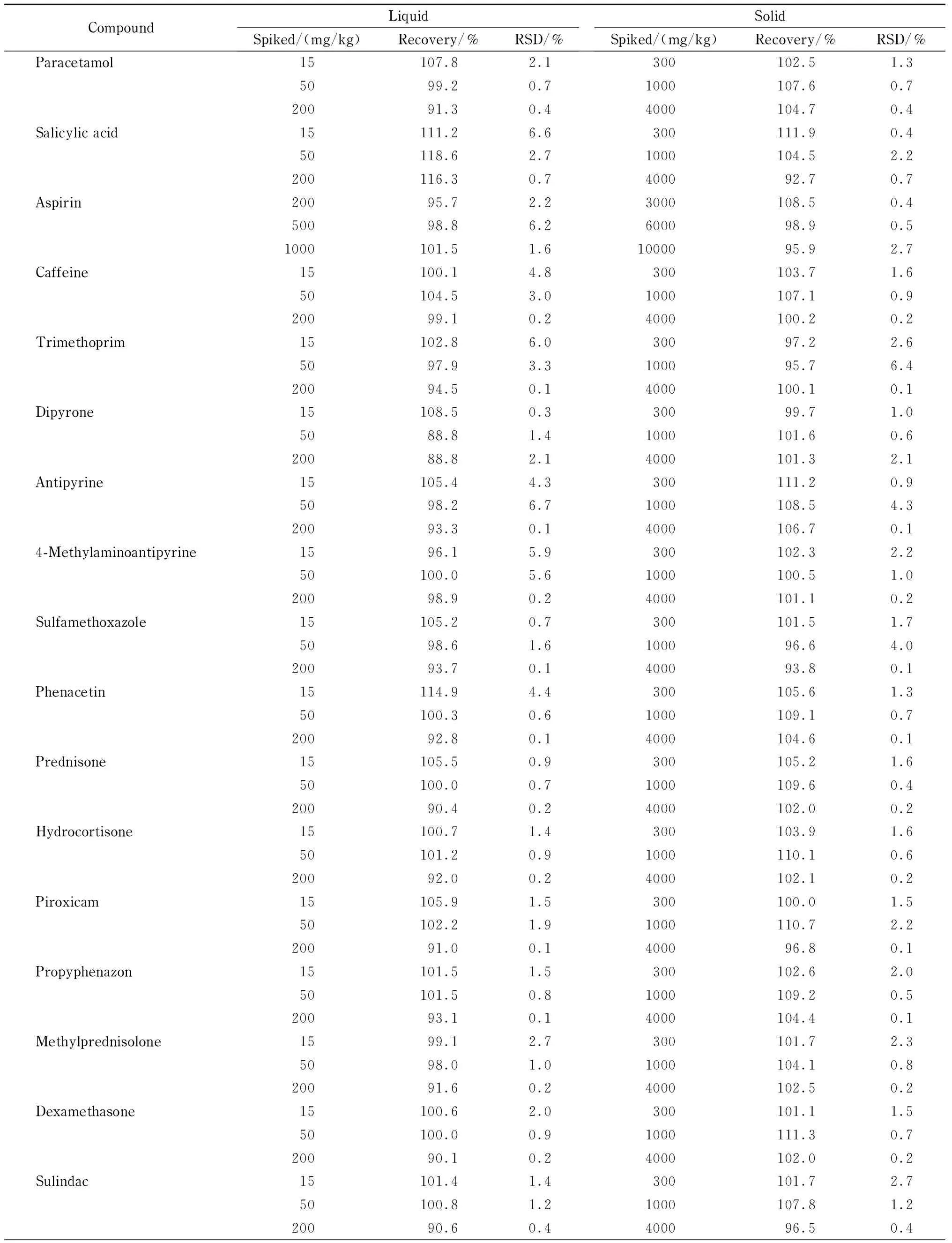
表 2 液体样品和固体样品中28种成分的加标回收率及RSD(n=6)Table 2 Spiked recoveries and RSDs of the 28 components in the liquid and solid samples (n=6)

表 2 (续)Table 2 (Continued)
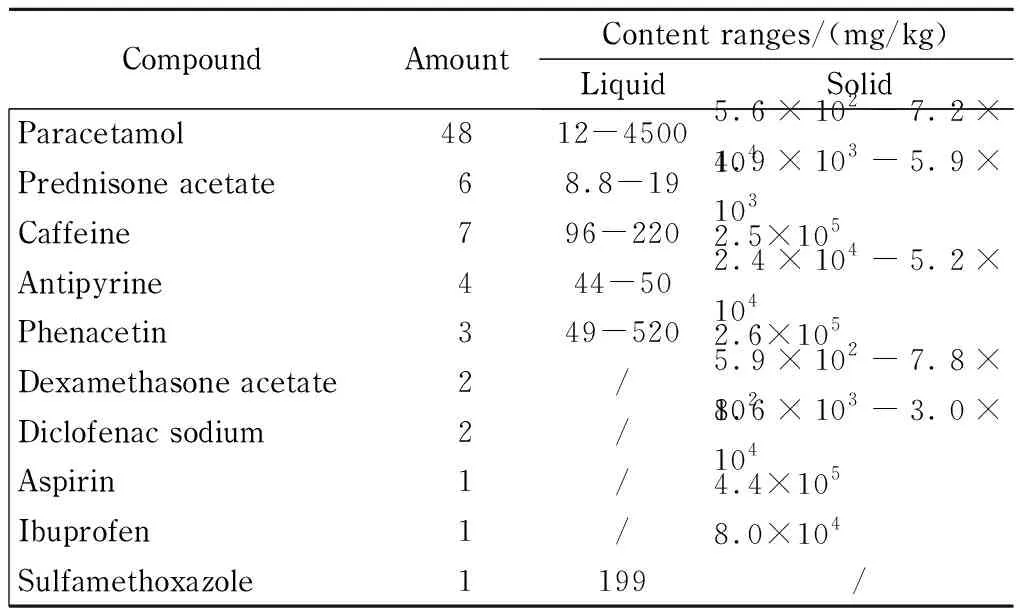
表 3 阳性成分的检出数量和检出含量范围Table 3 Detected amounts and detected content ranges of the positive compounds
/: not detected.
2.4.3植物内源性成分的判定
草本植物饮料原料多为药食同源的中药材,经煎制后成品的具体组分尚未明确。水杨酸为植物内源性激素,存在于柳树皮、烟草、黄瓜等植物中。456批样品中,没有单独检出水杨酸,1批样品中检出水杨酸和阿司匹林。经讨论,阿司匹林在水中易水解得到水杨酸,若两者同时检出,检出的水杨酸很有可能是由阿司匹林产生的。单独检测水杨酸意义不大,当只检出水杨酸时,不应将其折算为阿司匹林。本实验水杨酸检测的目的是辅助阿司匹林含量计算。
咖啡因是一种植物生物碱,存在于咖啡、茶及一些可可中,可作为食品添加剂用于可乐型碳酸饮料。然而,咖啡因也是一种中枢神经兴奋剂,可加强其他解热镇痛药物的疗效,是较常用的感冒药处方。456批凉茶中,有3批单独检出咖啡因,均为预包装产品;有4批同时检出咖啡因与其他解热镇痛药,为现场制售凉茶或凉茶粉。在预包装产品中单独检出咖啡因时,可参考其标签上的食品添加剂信息和食品分类,判断是否非法添加。若咖啡因与外源性药物同时检出,则有作为感冒药被加入的可能,其检出可以作为非法添加行为的一种提示。水杨酸、咖啡因的检出不代表非法添加行为的存在,应综合考虑各种因素,根据实际情况加以判定。
3 结论
本文考虑到草本饮料中非法添加药物的浓度水平和差异性,结合实际工作中液相色谱-质谱联用法出现假阳性的问题,建立了快速测定草本植物饮料中28种外源性药物和内源性成分的HPLC分析方法。该法优化了柱体系、流动相体系、提取溶剂等参数,并对易降解成分的计算、内源性成分的判定等进行讨论,操作简便,重复性高,可为草本植物饮料中非法添加药物的监督检验提供有力的技术支撑。
猜你喜欢
杂志排行

色谱的其它文章
- 全二维气相色谱-质谱分析煤油基吸热型碳氢燃料烃族组成
- QuEChERS-超高效液相色谱-串联质谱法测定不同植被类型土壤中11种甲氧基丙烯酸酯类杀菌剂
- 离子液体分散液液微萃取-超高效液相色谱-串联质谱法测定食品接触材料中全氟辛酸和全氟辛烷磺酸的迁移量
- 超高效液相色谱-串联质谱法快速测定鸡蛋中氟虫腈及其代谢物残留
- 固相萃取-高效液相色谱-串联质谱法同时测定牛奶和羊奶中莫奈太尔及其代谢产物残留
- Determination of 11 synthetic musks in imported seafood by solid phase extraction and gas chromatography-mass spectrometry