特发性高钙尿与骨质疏松的研究进展
2018-08-02张薰月综述冯正平
张薰月 综述 冯正平
重庆医科大学附属第一医院内分泌科,重庆 400016
特发性高钙尿是一种临床异质性疾病,并与骨量减少、肾结石形成密切相关。其多见于8-14岁的青少年或成人,多有遗传家族史,女性多于男性。目前已有大量研究表明,高钙尿主要有两大危害:一方面容易引起骨量减少、骨质疏松甚至骨折,另一方面高钙尿患者肾结石患病率明显升高,但其具体发生机制仍未完全阐明。本文对近年来高钙尿对骨代谢的影响以及与肾结石发生的可能机制的研究作一综述。
1 特发性高钙尿症的定义
Albright 在1953年首次提出“特发性高钙尿症”(idiopathic hypercalciuria,IH),特点是血钙正常而尿钙增高,需排除引起高钙尿的继发因素。此病儿童发病率约5.6%[1],肾结石复发患者的发病率为40%~60%[2]。
2 诊断标准及分类
过去根据性别差异将高钙尿诊断标准定为女性尿钙排泄> 250 mg/d、男性尿钙排泄> 300 mg/d。目前国际上公认将尿钙排泄量大于4 mg /kg·d作为诊断标准。根据具体发生机制的不同,IH 可分为肠吸收性(absorptive hypercalciuria,AH)、肾性(renal hypercalciuria,RH)、骨吸收性(resorptive hypercalciuria)3 类。
3 高钙尿临床后果
高钙尿患者易引起骨量减少、骨质疏松(Osteoporosis,OP),并增加骨折和肾结石风险,且具一定遗传易感性,而针对高钙尿的治疗同时可改善骨密度。已有多项研究证实IH 患者不论是儿童还是成人均常伴有BMD 降低及椎体Z值减少。Artemiukl[3]等对患有特发性高钙尿的儿童进行研究,发现有25.8%的患儿L1-4BMD 的Z值<-1,而在腰椎骨密度降低的人群中有50%患肾结石。Freundlich等[4]对21 名特发性高尿钙的儿童和他们无症状的母亲进行研究,发现8 名儿童和7 名母亲的骨密度下降,5 名母亲患高钙尿。骨密度下降的儿童的母亲腰椎BMD 显著下降。Letavernier E[5]对65个特发性高钙尿合并肾结石患者进行研究,发现该人群中35%有骨量减少,且与正常骨量人群相比前者呈现出更高的空腹尿钙。同时还发现,空腹尿钙与BMD呈负相关,空腹尿钙/肌酐比值大于0.25的人群发生低骨量的风险增加3.5倍。而在骨质疏松患者中,高钙尿患病率增高现象也普遍存在[6,7],Rull等[8]将两组均无肾结石病史的绝经后妇女分为骨质疏松合并骨折组及无骨质疏松组,发现OP组高钙尿发生率为40%,无骨质疏松组为18.8%, (P=0.04)。
针对高钙尿的治疗不仅可使尿钙排泄减少,同时可改善骨密度。一项随访六年的研究表明[9],针对高钙尿的枸橼酸钾和噻嗪类利尿治疗不仅使尿钙排泄从治疗前的5.0 mg/kg·d减少到2.6 mg/kg·d,同时腰椎骨密度的Z值也从治疗前的 -0.763±0.954 (平均值±标准差) 增加到-0.537±0.898 (P<0.0001)。也有研究认为是骨质疏松引起尿钙增高,高钙尿可能是骨转换增加的结果。
高钙尿与肾结石常伴随存在,并具一定遗传因素。Issler N等[10]的一项回顾性研究发现,322名泌尿系结石儿童患者中91名有高钙尿症,比例高达28.3%。另外,许多学者发现特发性高钙尿儿童中有肾结石家族史,证明IH与遗传有一定有关系。
4 高钙尿引起骨质疏松的机制
4.1 骨代谢异常
核因子κB 受体活化因子配体(RANKL) /核因子κB 受体活化因子(RANK) /护骨素(OPG) 通路激活可能是IH患者骨质疏松风险增高的主要机制之一。RANKL 由成骨细胞及骨髓基质细胞分泌, 与破骨细胞前体细胞或破骨细胞表面上的RANK 结合, 刺激前体破骨细胞分化, 促进破骨细胞成熟,引起骨吸收, 而OPG 为RANK RANKL 的诱饵受体,与RANK 竞争结合RANKL, 从而抑制破骨细胞分化、活化。在特发性高钙尿患者的骨组织学研究中发现,高钙尿患者骨组织表达RANKL 和OPG明显升高[11],骨吸收增加,由此引起骨量减少。

图1 RANKL/RANK/OPG通路Fig.1 RANKL/RANK/OPG pathway
另一方面,IH患者存在骨转换、骨吸收增加。骨形成标记物有骨碱性磷酸酶(BALP),骨吸收标记物有 β-I型胶原降解产物(β-crosslaps)和尿脱氧吡啶啉(DPD)。特发性高钙尿结石患者,骨代谢标记物不同程度的升高。一方面,IH患者骨吸收标志物升高,Arrabal 等[12]发现 β-crosslaps 与 尿钙呈正相关。张思伟等[13]发现rIH 患者尿DPD 升高。故认为骨吸收增加导致了BMD的下降。另一方面,骨形成标记物明显增加,Luna-Cabrera F[14]等发现,24小时尿钙排泄与BALP显著正相关。 成骨细胞活性增强, 则BALP分泌增加。认为IH 患者体内存在负钙平衡,骨钙丢失、矿化不良最终引起骨转换增高、BMD 降低,而骨形成标志物增加是骨转换增高,成骨细胞活性增强的结果。
4.2 遗传因素
既往研究发现,高钙尿存在一定的遗传易感性。对高钙尿人群的连锁分析和关联研究已经发现了许多与其相关的基因。
降钙素是调节骨转换和钙代谢的重要激素,而降钙素受体(caltitonin receptor,CTR)在骨骼上主要分布在破骨细胞及其前体细胞上。扫描人类CTR基因发现有11个多态性位点,其中一个位点的核苷酸序列1377处的C突变成T,从而使CTR基因产生CC(纯脯氨酸)、TT(纯亮氨酸)、CT(脯氨酸、亮氨酸杂交型)3种基因型,此为CTR基因型C1377T多态性。该单核苷酸突变影响到其与配体的结合并改变受体信号转导的特性,从而影响了靶细胞对降钙素的反应。杨奕等[15]对湖北地区76例特发性高钙尿患者及 126 例健康对照者外周血标本基因组DNA,检测并分析CTR 基因C1377T多态性分布,证明了其在该人群特发性高钙尿的发生中起重要作用。而近年一项Meta分析[16]证实:CTR基因型与腰椎、wards三角部位BMD有关联。故CTR基因型C1377T多态性可能成为高钙尿及骨量减少的桥梁,成为联系两者的遗传标志物。
瞬时感受器电位阳离子通道V5(transient receptor potential cation channel V5,TRPV5)是瞬时感受器电位(transient receptor potential,TRP)的一个亚家族成员,V亚家族命名源自最先发现的成员香草精(Vanilloid)受体1(VR1),该亚家族共有TRPV1-TRPV6六个成员。TRPV5主要表达于肾远曲小管和集合管,TRPV5基因编码TRPV5 蛋白,此蛋白作为上皮细胞表面钙通道,激活远曲小管对钙的吸收,故主要作用是对尿钙主动重吸收,参与钙环境稳态调节。TRPV5基因敲除小鼠模型对钙重吸收减少会引起高钙尿,而钙稳态主要靠肾脏、肠道及骨骼三个器官调节。在饮食来源的钙不变时,经肾脏排出的钙增多,为了保证血钙平衡,骨钙释放入血增加,故骨量减少、骨质疏松风险增高。由此得出结论,可引起TRPV5表达减少的因素会引起高钙尿及增加骨质疏松风险,目前在动物试验中已经发现了明确的突变基因。
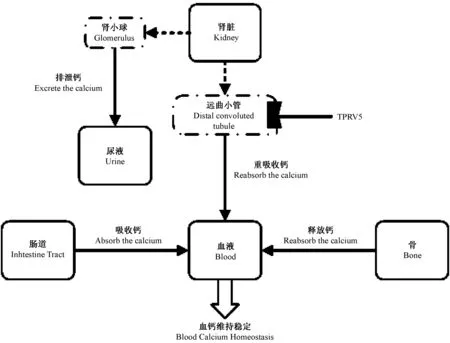
图2 血钙稳态的调节Fig.2 The regulation of calcium homeostasis
Loh[17]等认为TRPV5与常染色体显性遗传有关,他们发现TRPV5中第682位密码子T到C的突变引起TRPV5减少,显性基因是致病基因。与野生型(隐性基因)相比,杂合子和纯合子(显性基因)老鼠高钙尿发生率明显增多,肾远曲小管上对TRVP5选择性的表达减少与钙转运缺陷现象一致。他们还发现纯合子TRPV5(682P/682P)老鼠肾组织中细胞内钙结合蛋白减少,与TRPV 5-介导的肾重吸收钙缺陷现象吻合。故基因突变也是重要的机制之一。
近年对维生素D受体(VDR)基因的研究也颇多。1,25-(OH)2D3最重要的功能是调节钙、磷代谢,其主要是通过VDR介导的。人VDR基因存在单个碱基突变,这种突变在人群中有一定的分布频率,即单核苷酸多态性。IH相关的VDR基因多态性主要对应于4个限制性内切酶ApaI、BsnI、FokI、TaqI的酶位切点,对VDR基因多态性与IH相关性研究主要集中与上述酶位切点的多态性。Goknar N等发现[18]与正常尿钙的肾结石患者相比,高钙尿性肾结石患者中ApaI 中A等位基因的表达明显更高(P=0.036)。过去的研究已经证明, ApaIAA基因型组中血骨钙素水平高于Aa/ aa基因型,而骨钙素评估骨转换率及骨形成的灵敏和特异性指标。故ApaI AA基因型可能导致高钙尿和骨折疏松。但最近也有研究发现Apa1基因型 与骨质疏松无关[19]。目前关于ApaI 基因与高钙尿及骨质疏松的关系尚无定论,有待下一步研究。同样的,FokI基因与IH间的关系也有不同的结论。上述不一致的结论可能与种族等因素有关,不同人群IH易感性存在基因水平的差异。
高钙尿还可以通过VDR受体增多影响骨代谢。许多研究通过筛选正常雌雄大鼠(Sprague-Dawley rats,SD rats)中尿钙值的个体的交配繁殖并传代建立了遗传性高钙尿结石鼠(genetic hypercalciuric stone-forming rat,GHS rats),一般自第7代起,GHS表现出稳定的遗传特性:24 h尿钙排泄量显著高于正常对照组,同时BMD降低,而血钙、磷及 1,25-(OH)2D3是正常的,符合IH患者的临床特点。因此GHS鼠被公认为研究特发性高钙尿的理想模型[20]。维持钙平衡主要靠肠道、肾、骨三个器官,Frick 等[21]发现低钙饮食下,相同剂量的1,25(OH)2D3刺激,GHS组与对照组尿钙均有所增加,且GHS增加更为明显(P< 0.001)。已限定低钙饮食,而尿钙显著增加,故一定存在骨吸收增加。而Bai S等[22]发现高钙尿症伴尿石症大鼠小肠上皮和肾皮质VDR的表达无论是在mRNA还是在蛋白水平上均显著高于正常鼠,而且在微小剂量的 1α, 25(OH)2D3作用下, GHS 鼠VDR 的mRNA水平迅速升高 2.5-3.1倍。故认为尿钙在GHS组远超过SD组的原因是GHS大鼠VDR增多引起的对维生素D的高反应性。而在人群中,Favus[23]发现高钙尿结石患者外周血单核细胞中VDR的表达增高,但是否代表组织中VDR升高仍待进一步研究。
4.3 雌激素缺乏
雌激素可使OPG增加,RANKL减少,抑制骨吸收,是骨质疏松的保护因素。绝经后妇女雌激素骤然下降,易患骨质疏松[24]。近期对大样本含钙肾结石患者进行研究发现,与既往许多小型横向结果不同,Sakhaee K[25]发现:仅在没接受雌激素替代治疗的绝经后妇女中呈现出高尿钙、低骨量趋势,其余各组间尿钙排泄及骨密度均无显著差异。强调了雌激素在骨的完整性中的重要性,也再次印证了雌激素可能是引起骨质疏松、高钙尿的重要机制。绝经后妇女中患高钙尿率增高,一项研究[26]中发现204个雌激素缺乏的妇女中20%患高钙尿,尿钙排泄高于雌激素充足的妇女。而在1 572个老年男性骨质疏松骨折患者中没有发现高钙尿[27]。这都说明了雌激素缺乏的人群中尿钙排泄有所增加。故雌激素缺乏的妇女发生骨质疏松,除破骨细胞活性生成增加外,尿钙排泄增多也是重要的原因。
雌激素缺乏还可通过影响TRPV5的表达起作用。Dong X L等[28]发现切除卵巢的雌性大鼠,无论低钙还是高钙饮食下TRPV5 mRNA表达水平都比正常对照组明显减少。此研究从反面证实了雌激素对TRPV5及钙代谢的作用,与临床上妇女绝经前少见钙丢失、少见骨质疏松、IH发病少于男性的现象相符。提示雌激素缺乏可能还通过减少对TRPV5的表达从而减少钙重吸收起作用,一方面增加了尿钙排泄,另一方面也是骨质疏松的危险因素。由此得出结论,雌激素缺乏是高钙尿患者患骨质疏松的重要机制之一。
4.4 细胞因子增加
过去的研究已经发现BMD降低的高钙尿结石患者血、尿IL-1、IL-6、TNF浓度常有增高,普遍认为它们是通过与RNAKL结合,刺激破骨细胞增殖及功能表达,从而导致高钙尿及BMD降低的。但近年学者有不同的发现,并发现了一些新的细胞因子。Santos等[29]发现在IH患者中血、尿IL-1β, IL-6, IL-8,和TNF并没有增加。 而He D等[30]发现IH患者转化生长因子β1(TGFβ1)、骨形态发生蛋白(BMP2)和骨桥蛋白(OPN)的表达水平均高于对照组,解释了骨量减少。Santos 等[29]发现特发性高钙尿人群血清中单核细胞趋化因子-1(MCP-1)含量明显高于尿钙正常人群,而 MCP-1与BMD减少及雌二醇含量减少有关[31],故可能通过骨量减少及雌激素缺乏引起骨质疏松。
4.5 饮食
摄取过多的蛋白质会引起一系列代谢性酸改变,一方面抑制肾小管重吸收钙,从而钙排泄增加,引起IH;另一方面会使成骨细胞基质蛋白合成和BALP活性受到抑制,PGE2产生增多,成骨细胞表达RANKL 增加,促进破骨细胞的生成,导致OP。然而研究证明[32]高蛋白饮食尽管会加重尿钙排泄,但并不是引起尿钙排泄增加的主要原因。
5 高钙尿引起肾结石的可能机制
高钙尿可通过骨吸收增强、氧化应激损伤及遗传因素等多种机制引起肾结石,并且可能通过共同的通道引起骨量减少及肾结石。
总之,血钙平衡主要靠肠道钙吸收,肾钙的排泄及骨钙的再吸收维持,高钙尿的出现意味着三者维系的平衡被打破,从而引起骨代谢异常及泌尿系结石两大主要危害。近年来高钙尿受到人们的重视,而纠正高钙尿既能防止 BMD 的降低亦能预防泌尿系结石的发生,因此,对高钙尿引起骨质疏松、肾结石机制的研究临床意义大,而遗传因素可能是未来研究的方向。
