家族性慢性小管间质性肾病1例家系调查和基因筛查
2018-07-25徐海山唐建英许光辉
徐海山 唐建英 许光辉
有关家族性慢性小管间质性肾病的国内报道很少, 但是国内未查到慢性小管间质性肾病家系临床调查研究的相关文献。有研究发现, PAI-1、TIMP-1和Smad7基因与肾间质纤维化的发生有关, 但尚无这3个基因与常染色体显性遗传性肾小管间质肾病(ADTKD)相关报道。为此, 作者对临床上遇到的1例家族性慢性小管间质性肾病进行家系调查, 并初步探索是否存在上述3个基因异常。现报告如下。
1 资料与方法
1.1 一般资料 莆田学院附属医院临床诊治发现1例家族性慢性小管间质性肾病患者, 采集其家系, 成员总数为36例,累计4代。2016年和2017年2次进行家系调查, 共调查36例成员, 对其进行尿常规+沉渣镜检和肾功能电解质筛查, 对部分可疑成员进行尿蛋白成分分析以进一步诊断。将调查对象分为未受累者、患者和不明确者3种表型。有下列情况之一者视为未受累者:①家系中无亲缘关系的配偶, 亦视为未受累者;②年龄≥30岁, 尿检正常和肾功能电解质正常, 考虑该家系早发起病, 视为未受累者。有下列情况之一者视为患者:①肾脏病理证实慢性小管间质性肾病;②出现ESRD, 需要或已经进行替代治疗;③尿常规:对尿蛋白 (± ~ +)或尿比重异常或血清电解质异常或高尿酸血症者或血肌酐升高者作为疑似患者。下列情况之一者视为不明确者:①血肌酐升高, 且尿常规出现尿蛋白(2+~3+);②年龄<30岁, 尿检正常和肾功能电解质正常。所有受累和疑似患者均除外引起继发性原因, 如结缔组织病、反流性肾病、病毒感染、药物等。
1.2 方法
1.2.1 提取外周血细胞基因组DNA 在获取其知情同意后,采集了32例家系成员的外周血3~5 ml, 用SigmaDNA抽提试剂盒, 参照说明书操作步骤提取DNA, 经浓度和A 260/280比值测定合格后-20℃保存。
1.2.2 引物设计 部分外显子引物用primerprimier 5软件自行设计, 部分外显子引物来自参考文献。PAI-1、TIMP-1、smad7基因序列来自Genbank(NC_000007.14、NC_000023.11、NC_000018.10)。用相应外显子单侧PCR扩增引物进行测序。
1.2.3 PCR反应条件与体系 外显子采用反应体系为25 μl:10×PCR 缓 冲 液 2.5 μl, dNTPMixture (各 2.5 mM)2 μl,TaKaRaExTaq 酶 (5 U/μl)0.15 μl, 模板 DNA 30 ng。反应条件 :95℃预变性 5 min 后进入循环 :95℃变性 30 s, 退火 30 s(退火温度参照反应实际温度 ), 72℃延伸 45 s, 循环 36 次 , 最后72℃延伸 10 min。
1.2.4 PCR产物纯化与测序 按照QIAquickPCR产物试剂盒说明书收集纯化PCR产物, 然后用1%琼脂糖凝胶进行电泳估算其浓度, 纯化PCR产物以4℃保存。根据BIG dyeterminator测序试剂盒和ET-termi-nator测序试剂盒按照说明进行操作, 取适量纯化PCR产物, 用ABI 3730自动测序仪测序(美国PerkinElmer, Appliediosystem)。为了以保证结果的可靠性, 标本行重复和(或)双向测序。
1.2.5 碱基序列对比及SNP数据库搜寻 测序输出序列后,与模版序列匹配(BLASTbl2seq)比对, 以寻找突变位点;然后人工读序列图再次核实(Chromas 2.31)。如发现碱基变异,在SNP数据库搜寻以确认是否为单核苷酸多态性。新发现的碱基变异则以该家系中未受累者及其他健康人共10例为对照进行比对。
2 结果
2.1 家系的遗传方式 该家系成员均为汉族, 肾病家族史明确, 家系图可见图1。图谱分析显示该家系遗传方式为常染色体显性遗传, 即:①遗传和性别无关:男女均可发病,男女病情严重程度无明显差异;②患者父母中必有一方为受累者, 患者将异常基因传给子女的几率相等, 均为50%;③在连续世代中均每代均有成员发病。
2.2 家系的临床特征 家系共有4代, 成员总数49例, 调查成员36例, 其中3代连续发病。根据本研究确定的受累患者标准:共有患者5例, 未受累者12例。见图1。
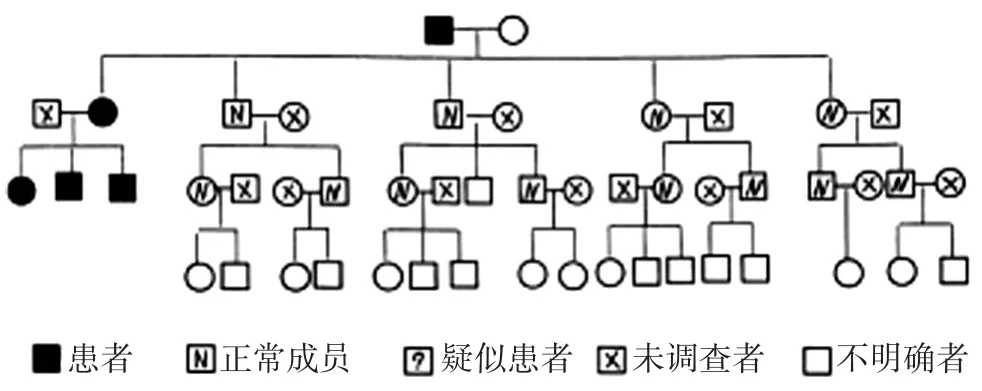
图1 患者家系图
2.3 先证者Ⅰ , 女性 , 21岁发现尿毒症 , 血液透析 3.5年后肾移植, 现在肾移植已6年, 目前生化全套、尿常规、血常规均正常。先证者Ⅱ, 男性, 先证者Ⅰ的大弟弟, 14岁发现也已经尿毒症, 血液透析1年后肾移植, 现在肾移植已10年,2017 年 12 月血肌酐 176.8 μmol/L、尿酸 416.3 μmol/L、尿素氮 13.5 mmol/L, 尿蛋白 ±。先证者Ⅲ , 先证者Ⅰ二弟弟 , 3 年前19岁首次体检发现血肌酐 136.6 μmol/L, 血尿酸446 μmol/L,尿蛋白±~1, 肾穿提示慢性小管间质疾病, 长期服用“尿毒清、金水宝、氯沙坦”2017年12月血肌酐200.8 μmol/L、尿酸 453.8 μmol/L、尿素氮 14.9 mmol/L, 尿蛋白 1+。三位先证者均无痛风和肾囊肿。先证者的爷爷和妈妈各自在30余岁时死于尿毒症。
2.4 家系先证者的基因筛查结果 家系先证者PAI-1、TIMP-1、smad7三个基因的所有外显子全部扩增成功, 产物纯化测序后比对未发现基因突变。
3 讨论
1971年国外报道了首例家族性慢性小管间质性肾病,后来这类疾病曾被命名为髓质囊性肾病、家族性间质性肾炎、遗传性质性肾病等。这类疾病命名混乱, 诊断标准不一, 不便比较和深入研究。因此, 改善全球肾病预后组织(KDIGO)最近将此类疾病命名为常染色体显性小管间质性肾病(ADTKD), 并发布了有关ADTKD的专家共识[1]。
ADTKD的病因尚未明确, 该共识认为ADTKD的发生主要与UMOD、REN、HNF1B、MUC14个基因有关, 也存在其他基因异常的可能。本病的临床表现十分接近, 无论是以上4个基因中哪个突变, 均是以肾脏浓缩稀释功能障碍为主要表现为;但是, 即使是同一个基因突变, 其发病年龄、病情严重程度、病情进展速度等均存在明显差异。本病进入ESRD 的年龄一般在 20~80 岁[2]。
ADTKD诊断标准:患者若有典型的间质性肾损害的临床表现, 同时具有明确的至少两代直系亲属中有类似的肾病病史且符合常染色体显性遗传模式的家族史, 就可以诊断本病。
有关ADTKD报道甚少, 仅有个案报道, 毛文丽等[3]报道1例;但是有关其家系调查国内报道尚无。作者研究的该例家系调查发现, 三代均有ESRD患者, 男女都有, 其中年轻1例患者肾穿明确是慢性小管间质性肾病, 据此符合ADTKD诊断标准。本家系都是非常年轻就发病, 很快进入ESRD, 未发现痛风和肾囊肿。
许多研究发现 , PAI-1、TIMP-1和 Smad7 基因与肾间质纤维化的发生有关[4,5]。但尚无这三个基因与ADTKD相关报道。作者研究发现此例ADTKD患者家族中PAI-1、TIMP-1和Smad7 基因并无突变, 据此作者认为这三个基因不是该本例家族性慢性小管间质性肾病的致病基因。ADTKD的治疗目前尚未有效的治疗手段, 需要更多的研究去探索其发病的机制和治疗。
