肝脏疾病细胞死亡方式及其研究进展
2018-07-20王涛王海久王志鑫任利任宾樊海宁
王涛,王海久,王志鑫,任利,任宾,樊海宁
(青海大学附属医院,西宁 810000)
细胞死亡是细胞生命的终点,亦是人体维持细胞总数的重要机制。肝脏作为人体内最大的消化腺,对人体的消化、代谢、内分泌调节等有重要作用,正常情况下,肝脏内存在细胞死亡与再生的平衡状态,外界刺激导致肝脏损伤,残余正常肝具有强大的再生能力。坏死、凋亡是人们较早认识的细胞死亡方式,近年来人们探索坏死与凋亡,对二者更为深入认识的同时,还发现了坏死性凋亡、焦亡、自噬、铁死亡等多种新的细胞死亡方式,这些新发现的细胞死亡方式在肝脏疾病的发生发展中有重要作用。现就目前坏死性凋亡、焦亡、自噬、铁死亡的形态学改变、发生机制及在肝脏疾病中的研究进展作一综述。
1 坏死性凋亡
传统观念认为坏死是一种不可控的细胞死亡方式,而凋亡是细胞内程序性死亡的结果,坏死性凋亡的出现颠覆了这一观念,2005年Degterev发现一种介于坏死与凋亡间的细胞死亡形式——坏死性凋亡,具有坏死形态学改变的同时又可被程序性调控。坏死性凋亡在形态学上与坏死类似,细胞膜破裂、细胞器肿胀、线粒体功能异常、膜的破裂导致细胞内大量成分外泄、周围组织的炎症反应,且不可被Caspase抑制剂抑制,但可被特异性抑制剂Necrostatin-1(Nec-1)抑制。目前对坏死性凋亡发生机制研究较深入的是肿瘤坏死因子(TNF)介导的死亡受体途径(图1)。
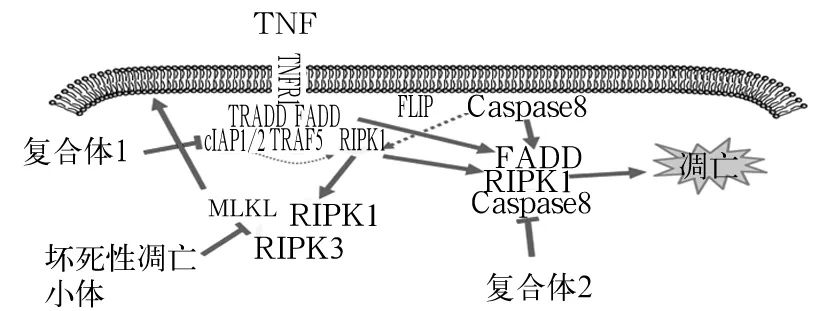
图1 坏死性凋亡中TNF介导的死亡受体途径
细胞表面存在诸多死亡受体,可与多种配体特异性结合引起细胞凋亡,如Fas、肿瘤坏死因子受体1(TNFR1)、DR3、DR4(TRAILR)、DR5、DR6、EDAR、NGFR等,此类受体的共同特征为在其胞质区有一段特殊氨基酸序列-死亡结构域,通过该死亡结构域可激活并特异性结合相应的衔接蛋白,引起相应的Caspase活化,导致凋亡发生[1]。当TNF结合于细胞膜上TNFR1后,TNFR1的胞内Fas相关死亡结构域(FADD)和肿瘤坏死因子受体相关死亡结构域(TRADD)募集下游受体交互作用蛋白(RIPK1)、TNFR相关因子5(TRAF5)、胞内凋亡蛋白抑制因子1/2(cIAP1/2)等多种下游分子,形成复合体Ⅰ,复合体Ⅰ中的cIAP可通过泛素化作用于RIPK1,促进细胞繁殖,同时去泛素化酶Cylindromatosis(CYLD)或去泛素化酶A20使复合体Ⅰ中的RIPK1发生去泛素化,其结果导致复合体Ⅰ裂解,并游离出RIPK1、TRADD。此时,胞内的FADD与游离的RIPK1再次结合,同时使无活性的FLIP-Caspase-8二聚体中的Caspase-8游离,生成激活状态的Caspase-8并结合于FADD、RIPK1上,形成复合体Ⅱa。当Caspase-8激活后可级联引起Caspase家族激活,导致凋亡,Caspase-8活性受到抑制时,RIPK1可磷酸化暴露其RHIM结构域,招募RIPK3,混合连接激酶结构域样蛋白(MLKL)同时使其磷酸化,形成RIPK1-RIPK3-MLKL小体,又称坏死性凋亡小体[2]。MLKL作为坏死性凋亡的执行者,可通过MLKL依赖性Ca2+内流和磷脂酰丝氨酸(PS)暴露于质膜外两种途径导致细胞膜完整性破坏,Ca2+的致死性流入由MLKL作用于胞膜瞬态受体电位阳离子通道亚族M成员7导致。ESCRT-Ⅲ组件是细胞膜保持完整性的重要组件,ESCRT-Ⅲ可保持细胞完整性及正常形态,磷酸化的MLKL作用于ESCRT-Ⅲ,导致胞膜形成富含PS的囊泡,破坏胞膜完整性,导致坏死性凋亡的发生[3]。
坏死性凋亡广泛存在于病毒性肝炎、酒精性肝炎、非酒精性脂肪肝炎、肝癌等多种疾病。Roychowdhury等[4]在酒精性肝炎模型中,使用Caspase抑制剂可抑制乙醇诱导的细胞凋亡,但不能完全抑制RIPK3表达及肝细胞变性等细胞损害,提示酒精性肝炎小鼠模型肝脏和酒精性肝炎患者肝组织活检标本中RIPK3过表达,进一步提示酒精性肝炎中坏死性凋亡的存在。血红素氧合酶-1(HO-1)是一种氧化应激诱导酶,具有抗凋亡和抗感染的作用,常用原卟啉钴(CoPP)诱导其在肝细胞中的表达,CORM-A1是一种CO释放颗粒,Bakytzhan等[5]在用CoPP和CORM-A1治疗酒精性肝炎模型后,发现HO-1和CO可改善酒精性肝炎的谷草转氨酶升高和炎性细胞因子表达,提示坏死性凋亡可能与氧化作用有一定联系,但目前其机制尚不清楚。Sanjoy等[6]通过高脂饮食干预小鼠模型,发现高脂饮食可增强RIPK3表达及MLKL磷酸化,加重RIPK3缺失小鼠的肝细胞损伤和脂肪变性及肝纤维化,RIPK3缺乏可下调MLKL磷酸化同时增强肝细胞凋亡的发生,以上结果提示Caspase-8缺乏使凋亡转化为坏死性凋亡,同时RIPK3缺乏亦可将坏死性凋亡转化为凋亡,RIPK3在保护肝细胞方面发挥重要作用,这可能与坏死性凋亡结合内环境中大量炎症因子,避免炎症放大,从而减轻损害有关。对乙酰氨基酚(APAP)作为一种解热镇痛药物,其常见并发症是肝毒性损害,长时间以来,人们用特异性抑制剂Nec-1减轻APAP继发的肝毒性损害,但最近研究发现,基因敲除RIPK3及MLKL小鼠模型并未起到减轻APAP继发的肝毒性损害作用,而基因敲除RIPK1小鼠模型却有明显抑制效果,此说明坏死性凋亡可能存在一种不依赖于RIPK3的独立信号机制,尚待进一步发掘[7]。
2 自噬
自噬是细胞清除自身错误蛋白质与受损细胞器的过程,近年来由于Yoshinori Ohsumi在自噬领域的重大贡献使其成为目前研究较多的细胞死亡机制。目前主要有两种分类方法,根据降解的底物分为选择性自噬与非选择性自噬,根据底物进入溶酶体的方式分为大自噬、小自噬、分子伴侣介导的自噬。大自噬的主要过程是自噬体形成后,包裹细胞质中受损的线粒体,细胞器及细胞内病原体,包裹后通过膜融合的方式与溶酶体结合,形成自噬溶酶体并降解包裹内容物,并再次利用。小自噬即胞质中的上述物质直接与溶酶体表面结合、凹陷,进入溶酶体内被降解的过程。分子伴侣介导的自噬是胞质中的分子伴侣与底物结合形成复合体,结合并转位至溶酶体内部,从而最终降解底物。自噬信号机制及种类繁多,且近年来不断完善,在此仅介绍人体中大自噬的基本发生机制。大自噬的发生机制主要包括欧米茄体的形成、自噬体的延伸、自噬溶酶体的形成、溶酶体的降解再利用四个基本过程(图2)。

图2 大自噬体的形成机制
2.1 自噬的开始 传统观念认为自噬发生的场所为内质网,近年来发现线粒体与自噬发生明显相关,研究组最近发现自噬体实际上很有可能起始于内质网和线粒体的交接处,并将该处交界的膜结构命名为线粒体相关膜(MAM)。雷帕霉素靶蛋白C1(mTORC1)可在感受到外界饥饿,氧化应激等刺激下,与unc-51样自噬激酶1(ULK1)复合体相互作用。机体能量充足的情况下,mTORC1处于高度活化的状态,可使ULK1复合体磷酸化,抑制自噬的发生,当机体能量缺乏时,ULK1复合体去磷酸化,诱导自噬发生[8]。ULK1复合体激活后可在自噬相关蛋白(ATG)的帮助下,定位于MAM,但其具体参与欧米茄体的形成机制尚不清楚。Ⅲ型磷脂酰肌醇激酶(PI3K)复合体可结合到内质网、线粒体的膜组分,并将脂分子中的磷脂酰肌醇(PI)转化成磷酸磷脂酰肌醇3(PI3P)[9],饥饿条件下,定位于内质网和高尔基体上的蛋白DFCP1可通过FYVE结构域结合PI3P,DFCP1被诱导后集聚成类似于“Ω”的结构,因此被称为欧米茄体。
2.2 自噬体的延伸 胞质中存在两个泛素化系统,ATG12-ATG5-ATG16复合物与微管相关蛋白质轻链3(LC3)-磷脂酰乙醇胺(PE)复合物[10],二者分别由ATG7、ATG10和ATG4、ATG7、ATG3介导形成,作用于欧米茄体,促进延伸,生成自噬体,其中LC3-Ⅱ是自噬体生成的生物学标志。
2.3 自噬溶酶体的形成 自噬体形成后包裹错误蛋白质,损伤线粒体等底物,再与溶酶体发生融合形成自噬溶酶体,这是自噬体成熟的标志,此过程中膜的融合是自噬体成熟的主要事件。该事件主要由突触相关蛋白SNAP29、突触融合蛋白STX17、突触囊泡相关膜蛋白VAMP8参与形成,自噬体上STX17通过胞质中的SNAP29作用于溶酶体上的VAMP8,从而导致膜的融合,最终形成自噬溶酶体。
2.4 溶酶体的降解再利用 自噬溶酶体形成后,通过水解酶将错误的蛋白质、核糖体等分解为氨基酸,并通过自噬溶酶体的透性膜转运至胞质,以供机体再利用。
自噬的发生机制较为复杂,涉及大量其他调节信号通路。目前自噬在肝脏肿瘤,酒精性肝炎,脂肪性肝炎,药物性肝炎等领域均有较大发展。Rubicon是一种自噬发生机制中Beclin1相结合的负性调节因子,Tanaka等[11]在试验中证实Rubicon在非酒精性脂肪性肝炎中过表达,而Rubicon基因敲除后的小鼠肝脂肪变性,内质网损害及自噬增加,此说明自噬可能参与了胞内脂滴的消除,自噬抑制在非酒精性脂肪性肝炎中起重要作用。Yongjun Tian通过HBV转染Atg5基因缺陷型小鼠,证实HBV DNA复制对自噬的依赖性。目前就其可能机制提出两种假说:①HBV通过一种HBV管理蛋白HBx与PI3K复合体结合;②HBV病毒表面蛋白SHB与LC3特异性结合。目前就两种可能机制尚无明确定论。自噬的主要作用是清除细胞体内氨基酸及脂肪酸,这种降解机制被认为支持癌症的生存和增殖。但研究表明自噬可支持非小细胞肺癌的发生[12],自噬的缺失亦可导致胰腺导管腺癌(PDAC)的发生。此说明自噬在肿瘤发生中扮演双重作用,研究[13]发现,在小鼠肝癌前体细胞中,p62(一种慢性肝病的标志蛋白)的积累通过激活mTORC1和NRF2来驱动HCC肿瘤发生。此意味着自噬在肝癌发生中其促进作用大于抑制作用,目前自噬的研究较多,越来越多的机制被阐明并用于肝脏疾病。
3 焦亡(Pyroptosis)
人们在探索细胞死亡的试验中发现一种Caspase-1特异性依赖的细胞程序性死亡,2001年,Brennan和Cookson将这种独特的程序性细胞死亡方式命名为焦亡(Pyroptosis)。其主要形态学改变与坏死及坏死性凋亡相似,表现为细胞质肿胀,细胞核固缩,染色质DNA断裂,焦亡不完全同于坏死及坏死性凋亡,在肿胀过程中,坏死及坏死性凋亡细胞肿胀过程中可保持细胞形态均匀增大,焦亡细胞在此过程中肿胀程度较前两种死亡方式明显减小,同时产生突出于细胞表面的多个膜泡样突起,最终导致细胞膜受力不均破裂[14]。目前焦亡的发生机制主要集中于经典途径与非经典途径两种方式(图3)。
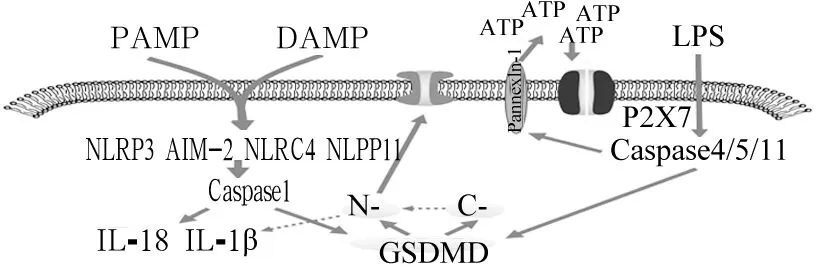
图3 焦亡发生的经典途径与非经典途径
3.1 经典焦亡途径 细胞在应对外界刺激时可通过模式识别受体(PRR)识别病原体相关分子模式(PAMP)与损伤相关分子模式(DAMP)。细菌、病毒、真菌等微生物的细胞壁成分及分泌物等均属于PAMP,来自机体外,当机体细胞受到外界刺激产生的某些可被PRR识别的有关成分被称为DAMP,由机体产生。PRR种类繁多,在焦亡发生中研究较为深入的有NLRP1b、NLRP3、NLRC4和AIM2四种炎症小体,其共同特点为在受到外界刺激激活后均可募集天冬氨酸特异性半胱氨酸蛋白酶-1前体(pro-Caspase-1),使其裂解形成有活性的Caspase-1。Caspase-1的作用主要体现在以下方面:①使Gasdermin D裂解为氮端(N-)与碳端(C-),Gasdermin D是Gasdermin蛋白家族中的一员,该蛋白家族目前功能除了与焦亡相关,其他功能尚未完全明确,但因其蛋白均含有Gasdermin结构域,故此命名。N-GSDMD具有成孔性及亲脂性,激活后可聚集于细胞膜表面形成非选择性通道,导致细胞渗透压失衡,同时可促进IL-18及IL-1β释放于细胞质,引起炎症反应。C-GSDMD的作用表现GSDMD未裂解时与N-GSDMD结合,抑制其细胞毒性,同时促进N-GSDMD水溶性,抑制其向细胞膜的聚集。②促进IL-18及IL-1β的成熟,IL-1β可上调体温调节中枢引起发热,属于内生致热源,同时趋化白细胞产生更多的炎症因子,放大炎症反应。IL-18主要是促进T细胞及巨噬细胞活化,促进炎症反应[15]。
3.2 非经典焦亡途径 不同于经典焦亡途径,细胞质中存在一类可直接识别革兰阴性菌脂多糖(LPS),而不依靠模式识别方式的天冬氨酸特异性半胱氨酸蛋白酶,其主要包括Caspase-4,5,11,其中Caspase-11主要存在于小鼠中,Caspase-4,5存在于人类。以上三种酶具有相似的三种功能:①裂解GSDMD介导细胞膜破裂,裂解方式及作用上文已阐述;②Caspase-4,5,11均不能直接加工IL-18、IL-1β前体,但在NLRP3介导的焦亡中可诱导低水平的IL-1β释放,目前其具体机制尚不清楚,可能与间接促进经典焦亡途径有关;③激活胞膜上的泛链接蛋白1,释放ATP,激活胞膜上的嘌呤P2X7受体,形成通道,最终介导焦亡发生[15,16]。
焦亡是一种与炎症反应相关细胞死亡方式,与肝癌、病毒性肝炎、酒精性肝炎、寄生虫感染甚至中暑后继发的肝损害有密切联系。2017年Khanova等[17]通过RT-PCR、免疫印迹法、免疫组化、酶联免疫吸附试验(ELISA)对酒精性肝炎小鼠模型及患者进行了焦亡相关关键因子的测定,并证实了Caspase-4,11介导的非经典焦亡途径存在于酒精性肝炎中。Beier等[18]证实GSDMD-/-小鼠在蛋氨酸-胆碱缺乏饮食喂养后未发生脂肪性肝炎与肝纤维化,可能是由于GSDMD在非酒精性脂肪性肝病(NAFLD)中能促进肝脏脂肪生成、炎症及焦亡,由此推测GSDMD的激活是非酒精脂肪性肝炎(NASH)与NAFLD重要的过渡点及潜在的治疗靶点。Kofahi等[19]在试验中发现HCV感染肝细胞中的Caspase-1水平明显升高,在Caspase-1特异性抑制剂Z-WEHD-FMK处理后可避免50%以上的HCV感染肝细胞死亡,该实验证实了焦亡是丙型病毒性肝炎继发细胞死亡的一种重要形式。在肝癌方面,Chu等[20]通过体外细胞试验证实了肝细胞肝癌(HCC)细胞中Caspase-1呈低表达状态,黄连素可通过上调Caspase-1蛋白表达抑制癌细胞的生存能力和迁移能力,Caspase-1特异性抑制剂Ac-YVAD-CMK可抑制黄连素的治疗效果,说明HCC的发生发展与焦亡的下调有关,此可能为临床HCC的治疗方式提供新方向。肝损伤是夏天中暑后最常见的并发症,亦是中暑最常见的致死原因,Geng等[21]在中暑的小鼠模型中发现Caspase-1高表达及NLRP3炎症小体,IL-1β的激活,细胞焦亡的形态学改变,该焦亡过程呈现高迁移率族蛋白1(HMGB1)依赖性,且NLRP3、Caspase-1、HMGB1三者的抑制剂均可抑制中暑继发的肝损害,但中暑以外的其他疾病引起肝损害是否依赖HMGB1尚无报道。焦亡在肝细胞损害发生发展中有重要作用,既可引起细胞释放炎症因子,增强免疫功能,抑制癌细胞增生,又可能因过度放大炎症反应引起机体损害,对焦亡涉及肝脏多种类型病变,进一步研究肝脏疾病的治疗大有裨益。
4 铁死亡
铁死亡是一种不同于凋亡、坏死、焦亡、自噬的全新细胞死亡方式,主要以铁稳态失衡、活性氧(ROS)产生为主要特点,且可被铁螯合剂Erastin抑制,故此,Dixon等将该死亡方式命名为“铁死亡”。其形态学改变主要是线粒体体积缩小,双层膜密度增加,线粒体嵴减少或消失。目前对铁死亡的研究还相对较少,其发生机制的研究主要集中于铁稳态失衡及谷胱甘肽过氧化物酶4(GPX4)活性下降两方面。
铁死亡的中心环节为Fenton反应,在该反应中Fe2+离子将H2O2和脂质过氧化物转化为ROS,自身被氧化为Fe3+,而GPX4在还原型谷胱甘肽(GSH)辅助下将H2O2和脂质过氧化物分别转化为H2O和相应的醇。铁死亡过程中ROS的主要作用是生物膜氧化损伤。膜脂富含多不饱和脂肪酸(PUFAs),PUFAs与ROS有较高的亲和性。因此,铁代谢产生的ROS可使生物膜氧化损伤,引起细胞死亡。
机体内的循环铁主要与转铁蛋白结合,以Fe3+存在,Fe3+通过膜蛋白转铁蛋白受体1(TFR1)进入细胞,并存在于内体中。在内体中,Fe3+被铁氧化还原酶(STEAP3)还原为Fe2+。最后,Fe2+转运体1(DMT1)介导Fe2+从内体中释放到细胞质中的动态铁库中。过量的铁储存在铁蛋白中,铁储蛋白复合体由铁蛋白轻链(FTL)和铁蛋白重链(FTH1)组成。铁输出由膜蛋白铁转运蛋白(SLC11A3)介导[22]。该机制形成了细胞质内铁离子相对稳定,铁稳态失衡后铁载量过大,这是铁死亡形成的重要因素。GPX4可竞争性抑制ROS的产生,当其活性受到抑制时,变相的敏化了铁死亡的发生。细胞表面存在名为systemXC-异二聚体,该二聚体由SLC7A11和SLC3A2组成,其主要作用为促进胱氨酸的吸收,排出谷氨酸,从而促进谷胱甘肽的合成,而谷胱甘肽是GPX4必需的辅助因子,当其合成受阻,导致GPX4活性降低,细胞抗氧化能力降低,ROS堆积,从而促进铁死亡的发生[23]。
目前铁死亡在肝脏方面疾病的应用主要在肝细胞肝癌(HCC)及药物性肝损伤两方面,索拉菲尼是目前惟一能提高晚期HCC患者生存率的口服药物,其主要机制为诱导HCC细胞凋亡、自噬等方式死亡,但最近Sun等[24]证明索拉菲尼和Erastin均能诱导肿瘤细胞发生铁死亡达到抗肿瘤疗效,该研究证实HCC细胞存在p62-Keap1-NRF2通路,通过p62调节Keap1降解,进而激活NRF2转入核内,上调铁和ROS代谢相关基因从而发挥抵抗铁死亡的作用。索拉菲尼可抑制NRF2活性,使HCC细胞对铁死亡更敏感,达到抗肿瘤效果。对乙酰氨基酚(APAP)引起的药物性肝损伤,APAP一方面破坏细胞内溶酶体,将Fe2+释放入胞质中,导致氧化应激。溶酶体的降解导致大量自由铁释放进入细胞质,从而敏化铁死亡[25]。另一方面,APAP与肝脏中蛋白质或非蛋白质的巯基结合,在毒性剂量时,细胞质和线粒体中含巯基的GSH严重耗竭,降低GPX4活性,导致肝细胞死亡。另外Carlson等[26]通过小鼠证实维生素E喂养GPX4缺乏小鼠模型可延缓小鼠生存时间,其机制可能与维生素E抑制有害的脂质氧化反应有关。目前铁死亡作为一种新的细胞死亡方式,其发生发展机制涉及肿瘤、细胞变性等多个方面,可为临床治疗提供新的靶点及方法,但目前在肝脏方面的研究还相对减少,有待进一步深入研究。
综上所述,随着对细胞死亡认识的不断加深,肝脏疾病中存在多种细胞死亡方式,其参与疾病的发病、炎症免疫等,亦涉及多种药物引起肝细胞损害及抗肿瘤效应,但目前具体机制尚不明确。进一步探究细胞死亡在肝脏细胞中的运行机制可为预防肝病发生,寻找新的治疗靶点,减轻肝脏损害等提供新的思路。
