UPLC-MS/MS快速检测动物源性食品中多种兽药残留1
2018-07-13翟莉芳刘战花
翟莉芳,刘战花
(惠州市农产品质量安全监督检测中心,广东 惠州 516008)
随着现在生活水平的逐渐提高,动物源性食品在人们日常饮食结构中所占比例逐步增大,需求量也逐渐增大.但是因为兽药(含药物饲料添加剂)在畜禽产品中的广泛应用,导致动物源性食品中的兽药残留的问题引起了人们的热切关注的社会焦点[1].为保证动物源性食品的卫生安全,我国已于2002年颁布法规在畜禽养殖中禁限用多种药物[2],但至今这类药物违法添加事件仍屡禁不止.
国内标准方法对动物源性食品的检测多局限于单一种类,如动物源性食品中β-受体激动剂[3]、动物源性食品中氟喹诺酮[4]、动物源性食品中磺胺类药物[5]. 不能够通过一次前处理,检测多种类的兽药残留,本实验通过一次前处理,能完成β-受体激动剂、氟喹诺酮、磺胺类药物的同时检测,节约了大量的检测分析时间.而且使用了新型的强效除脂型Oasis PRiME HLB固相萃取柱,能将动物源性食品中含有的大量脂肪、蛋白质、磷酸酯等杂质去除,不仅节约的检测成本,还提高了检测效率.
兽药残留检测常用的方法有酶联免疫法(ELISA)[6-7]、气相色谱-质谱联用(GC-MS)[8]、液相色谱法[9-10]、液相色谱-串联质谱联用(LC-MS/MS)[11]等.ELISA法虽然快速、方便,但在实际运用中假阳性率较高,不能实现在线检测;GC-MS在检测时需进行衍生,且前处理较繁琐;LC-MS/MS以其特异性强、高灵敏度,成为了现阶段兽药残留检测的首选方法.本研究只通过简单的提取后,固相萃取法对样品进行前处理,通过高选择性的超高效液相色谱-串联质谱(UPLCMS/MS)对处理后的样品进行测定,研制出了UPLCMS/MS快速测定动物源性食品中多种兽药残留的方法,为今后多残留检测方面的研究提供必要的技术支持.
1 实验部分
1.1 仪器、试剂与材料
主要仪器:UPLC-MS/MS 6460超高效液相色谱串联质谱仪(美国安捷伦公司);SHA-B水浴恒温振荡器(金坛市精达仪器制造厂);MS3涡旋振荡器(德国IKA公司);TDZ5-WS多管架自动平衡离心机(湘仪离心机仪器有限公司);N-EVAP TM 111恒温水浴氮吹仪(JNC公司);BS224S电子天平:感量0.1 mg(德国赛多利斯公司).
主要试剂与材料:乙腈、甲醇:色谱纯(德国Merck公司);甲酸、乙酸铵:分析纯(天津市广成化学试剂有限公司);固相萃取柱:Oasis PRiME HLB(60 mg 3 cc,Waters公司)微孔滤膜:0.22 μm(天津市津腾实验设备有限公司);25种兽药标准品均购自德国Dr.Ehrenstorfer,纯度均大于95%.
标准工作液:准确称取适量25种标准品,用甲醇分别溶解成100 μg/mL的标准储备液.
混合标准工作溶液:分别准确吸取1.0 mL的各种标准储备液至100 mL容量瓶中,用甲醇稀释至刻度.
1.2 仪器条件
1.2.1色谱条件
UPLC-MS/MS:色谱柱:C18,100 mm×2.1 mm,1.8 μm;柱箱温度:40 ℃;进样体积:8 μL;流动相:A相:0.2%甲酸水溶液(含5 mmoL/L乙酸铵),B相:0.2%甲酸甲醇;梯度洗脱程序:见表1.
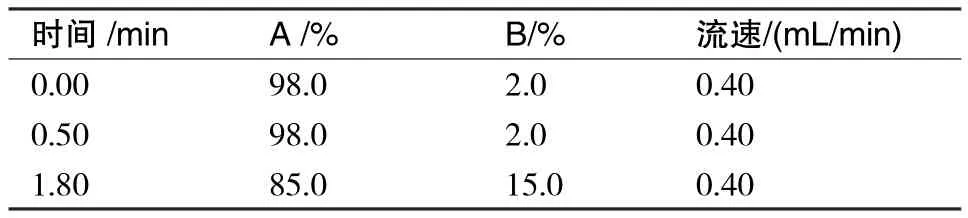
表1 UPLC-MS/MS梯度洗脱程序
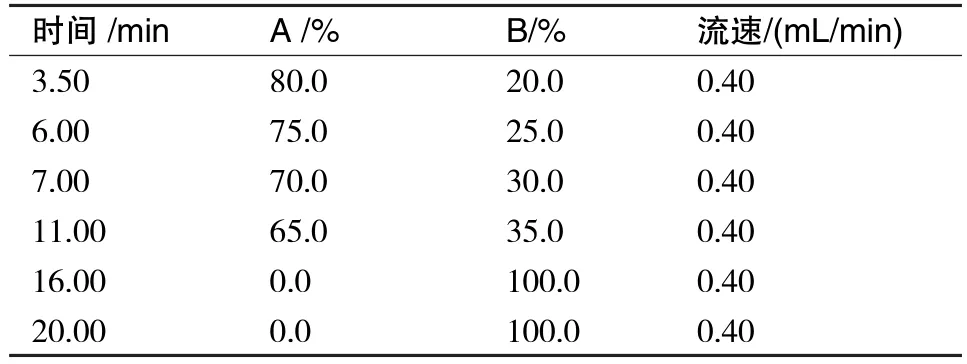
(续表1)
UPLC-MS/MS:离子源:ESI,多反应监测(MRM),正离子模式;干燥器温度:280℃;干燥器流量:9 L/min;雾化气:40 psi;鞘气温度:350 ℃;鞘气流速:11 L/min;20种兽药质谱测定的离子对、碰撞能量及保留时间如表2所示.按该质谱条件进样检测.
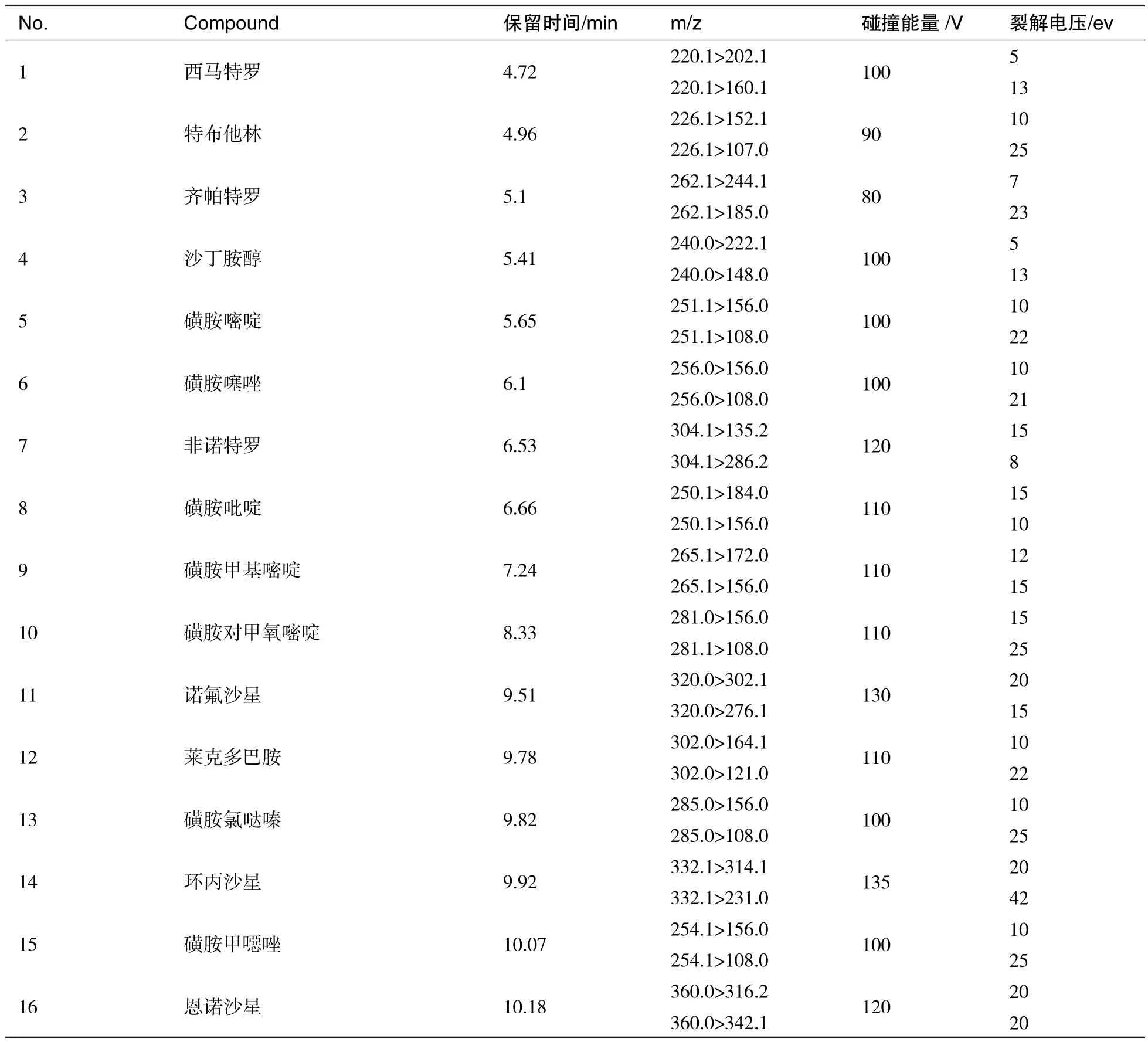
表2 25种兽药的保留时间、离子对、裂解电压和碰撞电压
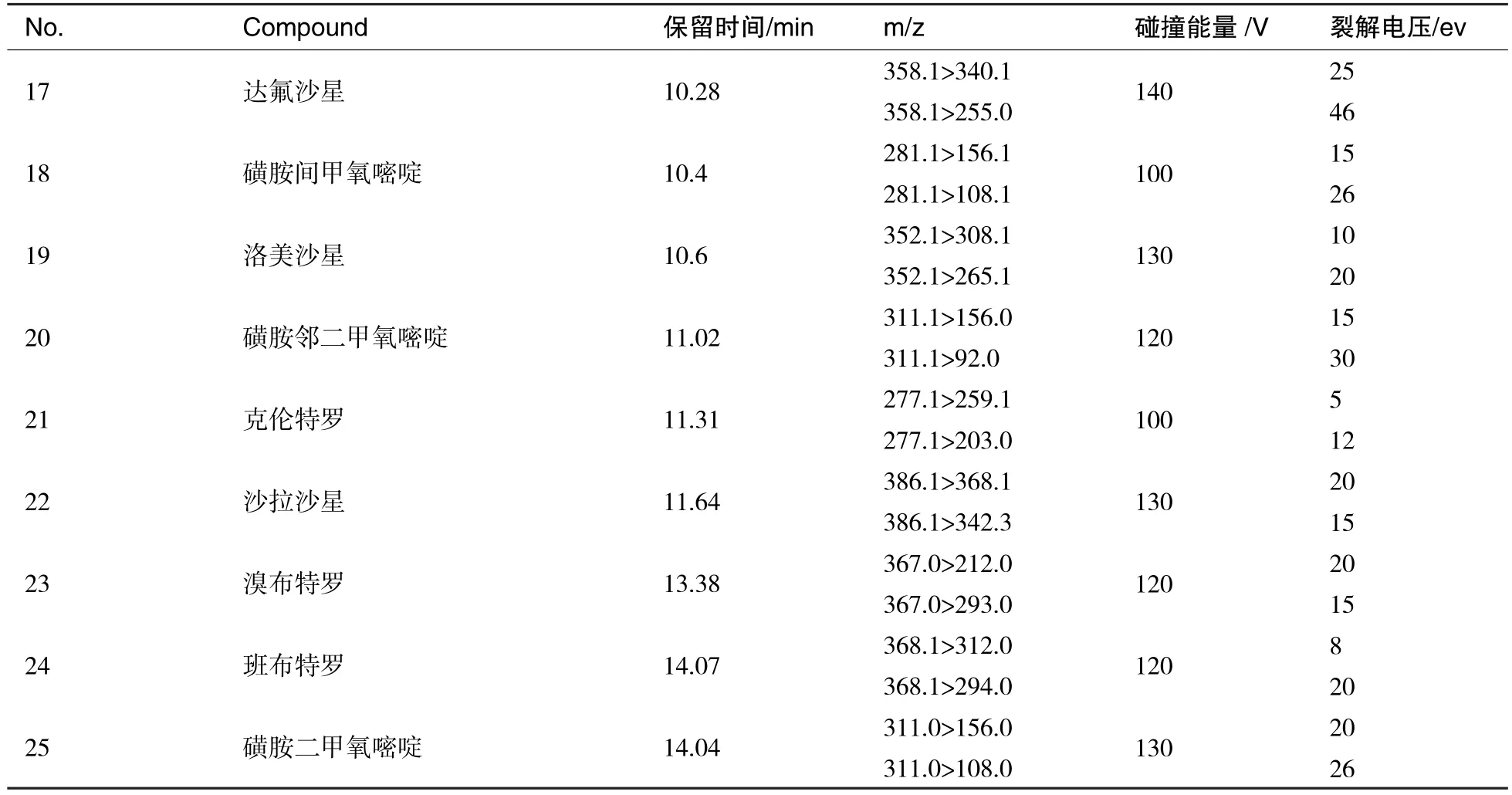
(续表2)
1.3 前处理方法
称取2.5 g组织匀浆样于50 mL塑料离心管,加入10.0 mL 0.2%甲酸乙腈水溶液(8:2),涡旋混合30 s,置于摇床上30 min,在10000 r/min下离心5 min,取上清液通过Oasis PRiME HLB固相萃取柱,收集滤液约2 mL,过0.22 μm滤膜,供UPLC-MS/MS测定.
2 结果与讨论
2.1 色谱行为

图1 UPU图1.UPLC-MS/MS中25种兽药混合标准溶液的总离子流程图
2.2 净化条件的确定
对于动物源性食品,传统的净化方法是使用正己烷去除脂类,但不能将动物源性食品中的蛋白质及磷脂除去,仍需要进一步净化.也有使用C18、MCX、SCX等固相萃取柱对样品溶液进行净化,但需要对萃取柱进行活化、上样、淋洗、洗脱等过程,步骤多,耗时长,也消耗大量的溶剂.本实验采用了Oasis PRiME HLB固相萃取柱进行简单、快速的净化,省去了活化、淋洗、洗脱等过程,还能有效的去除动物源性食品中脂肪、磷脂和蛋白质.研究还表明Oasis PRiME HLB小柱对25种兽药吸附很少,过柱前后,25种兽药的响应值变化不大,Oasis PRiME HLB固相萃取对样品中目标物造成的损失很小.
2.3 提取剂的选择
由于选用了特殊填料的Oasis PRiME HLB固相萃取柱对样品进行净化,省去了有机溶剂洗脱的过程,为了保证净化液能直接用于UPLC-MS/MS检测,因此选用了有机溶剂与水的混合溶液对样品进行提取.乙腈作为万能提取剂,常和甲酸等溶剂混合,配置成酸性乙腈被用于动物源性食品中提取兽药残留.但纯乙腈不利于杂质的去除,也不适用于仪器检测.而加入一定比例的水,不仅有利于从样品中提取目标物,还有利于减慢过柱速度,使杂质更好的被去除,使净化液更适用于仪器直接检测.因此,本实验比较了0.1%甲酸的乙腈水溶液(9:1)、0.1%甲酸的乙腈水溶液(8:2)、0.1%甲酸的乙腈水溶液(7:3)、0.2%甲酸的乙腈水溶液(9:1)、0.2%甲酸的乙腈水溶液(8:2)、0.2%甲酸的乙腈水溶液(7:3)等6种提取剂的提取效率.结果表明,当乙腈与水的比例为9:1时,仪器检测是目标峰出现拖尾.当乙腈与水的比例为7:3时,提取效果较差,添加回收率较低.0.2%甲酸的乙腈水溶液(8:2)不仅能很好的从样品中提取目标物,还能较好的去除杂质的干扰,且目标峰不会出现拖尾,更适合仪器检测.因此本实验选择了0.2%甲酸的乙腈水溶液(8:2)作为提取剂.
2.4 基质标准曲线、线性范围、检出限、定量限、回收率及精密度
为降低基质效应影响,保证检测结果可靠准确,采用基质标准曲线进行定量. 分别取20 μL、40 μL、80 μL、160 μL、320 μL兽药标准工作液,在50℃水浴下氮吹至干,加入1.0 mL对应基质空白溶液定容,涡旋混匀后,按照上述仪器条件进行测定,以外标法绘制标准曲线.25种兽药的线性相关系数r均大于0.996,表明25种兽药分别在浓度为20.0 ng/mL~320 ng/mL之间有良好的线性关系.
以3倍信噪比(S/N=3)为方法检出限(Limit of Detection,LOD),以10倍信噪比(S/N=10)为定量限(Limit of Quantification,LOQ),25种兽药的检出限为0.006 μg/kg~0.08 μg/kg,定量限为0.02 μg/kg~0.26 μg/kg.25种兽药的相关系数、检出限、定量限见表3.
在猪肉、猪肝、鸡肉样品中分别添加0.030、0.30 mg/kg混合标准溶液,按上述前处理方法进行处理,平行测定6次,得到平均回收率及精密度,各兽药的平均回收率为70.4%~90.6%,相对标准偏差(n=6)为5.3%~12.3%,结果见表4.

表3 25种兽药的相关系数、检出限、定量限
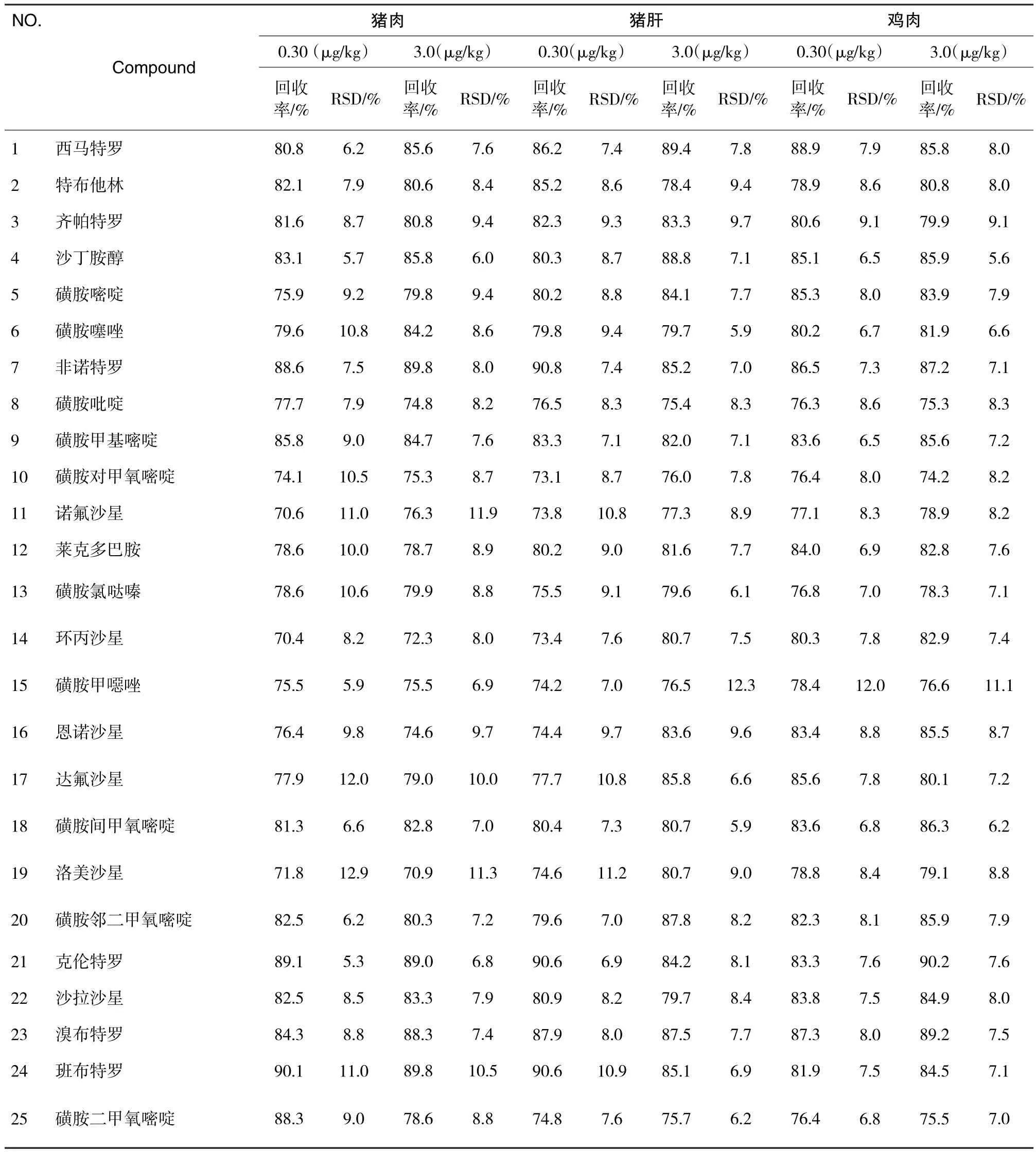
表4 精密度与回收试验结果(n=6)
2.5 实际样品测定
按实验方法对50批次的猪肉、猪肝、鸡肉样品进行测定,其中在一个批次的猪肝中检测5.7μg/kg的磺胺二甲氧嘧啶,在一个批次鸡肉样品中检测1.2μg/kg的恩诺沙星,均未超出国家限定范围,均为合格样品.
3 总结
本实验研究采用酸性乙腈水溶液对动物源性食品进行提取,Oasis PRiME HLB固相萃取柱对样品进行简单、快速的净化,建立了超高效液相色谱-串联质谱同时快速测定动物源性食品中25种兽药残留量的方法.25种兽药的回收率为70.4%~90.6%,相对标准偏差在5.3%~12.3%,检出限为0.006~0.08 μg/kg.该方法简单、快速、准确灵敏等特点,适用于动物源性食品中多种兽药残留量的测定.
