尼罗罗非鱼1号染色体分离及文库构建方法研究
2018-07-13李大宇邹芝英祝璟琳
耿 青,肖 炜,李大宇,邹芝英,祝璟琳,杨 弘
(1.南京农业大学无锡渔业学院,江苏无锡 214081;2.中国水产科学研究院淡水渔业研究中心,农业部淡水渔业和种质资源利用重点实验室,江苏无锡 214081)
尼罗罗非鱼(Oreochromisniloticus)原产于非洲,已成为我国主要养殖的淡水鱼类之一。养殖过程中尼罗罗非鱼的雄鱼生长速率显著高于雌鱼,引起研究学者对罗非鱼的性别调控的广泛兴趣。遗传学研究发现,罗非鱼的有丝分裂中期分裂相中存在一对中部着丝粒染色体,显著大于其他染色体对,并命名为1号染色体,推测1号染色体可能存在性别决定基因[1-3]。1998年Kocher等[4]构建了尼罗罗非鱼第一代遗传连锁图谱,后续研究表明,图谱上的部分分子标记与罗非鱼的性别连锁相关[5]。
目前,染色体的分离手段主要分为两种:玻璃针分离法和激光切割法。上世纪80年代,研究者首次运用显微玻璃针分离法来获得果蝇唾腺染色体特异区域,并成功克隆报道[6]。这项技术打开染色体分离技术在动植物遗传学应用研究的大门。同时,显微玻璃针分离技术对操作人员的实验操作要求较高,一定程度上限制该技术的进一步推广应用。近年来,激光微束切割仪的出现,研究者创造出利用激光微束,通过计算机控制轨迹,切割目标染色体的方法[7-8]。将该方法与PCR技术结合,推动单染色体扩增及文库构建的快速发展。目前,该技术相继在人[9]、小麦[10]、大麦[11]、棉花[12]、杨树[13]、山羊[14]等多种动植物的染色体显微分离和文库构建的研究中得到应用,但其在鱼类研究中的相关报道很少。本研究利用激光显微切割法收集尼罗罗非鱼的1号染色体,通过特异性酶切、LA-PCR体外扩增、电泳回收、连接转化、感受态细胞培养等步骤优化建立尼罗罗非鱼1号染色体微克隆文库的方法。为从尼罗罗非鱼1号染色体上筛选特异性性别相关标记,开展分子辅助育种提供技术支撑。
1 材料与方法
1.1 材料
LMD 6000显微操作系统为 Leica 公司产品,PHA、秋水仙素购自Sigma公司。蛋白酶 K、Sau3AI、T4连接酶等试剂均由TaKaRa公司提供。100 g左右的XY型尼罗罗非鱼由中国水产科学研究院淡水渔业研究中心南泉养殖基地提供。
1.2 方法
1.2.1染色体标本的制备
参考人的植物血细胞凝集素(PHA)体内注射法[15],按鱼体重以10 μg/g的量向一条XY型尼罗罗非鱼的胸鳍基部注射PHA(PHA为冻干粉制剂,以1 mg/mL的浓度溶解于灭菌的0.8% NaCl溶液中)28~30 h后,按鱼的体重以2 μg/g 的剂量注射秋水仙素溶液(秋水仙素为冻干粉制剂,以0.2 mg/mL的浓度溶解于灭菌的0.8% NaCl溶液中),1.5~2 h后将实验鱼解剖取出头肾组织,置于盛有0.8% NaCl的培养皿中,清洗2次。在100目的过滤筛网上研磨头肾组织,加入8 mL生理盐水过滤冲洗后移入15 mL的离心管中,用吸管反复吹打至细胞分散,制成细胞悬液。将细胞悬液以1 000g的转速离心 5 min,弃上清,收集底部沉淀。低渗处理:加入经37 ℃ 预温的0.75 mol/L KCl溶液低渗处理30 min,使细胞膨胀。再次将细胞悬液以1 000g的转速离心 5 min,弃上清,收集底部沉淀。预固定、一次固定和二次固定,按照普通核型制备条件进行[16]。制片:将预冷的POL膜载片放置成45 ℃角,从1 m高处滴片,每片滴加悬液2~3滴,再将膜载片放入70 ℃烘箱中放置30 s,干燥后用10%姬姆萨染色液染色30 min,然后用蒸馏水轻轻冲洗膜片,室温晾干后于-20 ℃保存。
1.2.2目标染色体的显微分离与收集
在LMD 6000激光显微镜的40倍物镜下,找到60个分散良好的分裂相,依次做好定位标记。将物镜调150倍油镜后,通过定位寻找到1号染色体,激光照射消除周围染色体和杂质,从中选出10对完整的1号染色体切下,保留于20 μL反应缓冲液(5 g/L蛋白酶K,1 ×T4 DNA连接酶缓冲液配制)的0.2 mL PCR管中。12 000g离心30 s,-20 ℃存放待用。
1.2.3蛋白酶K的消化与Sau3AI接头的制备
将含1号染色体的0.2 mL PCR管置于37 ℃下温育2 h,使蛋白酶K充分消化去蛋白,然后置于75 ℃下20 min灭活蛋白酶K。参考Chen等[17]的接头的制备方法,将100 μmol/L的19mer和23mer各1 μL加入到含58 μL dH2O的PCR管中,在PCR仪中90 ℃变性 2 min,55 ℃退火20 min,冷却后加dH2O 140 μL,浓度为50 ng/μL.取10 μL加入90 μL 20 mmol/L ATP(1 ×T4 DNA连接缓冲液配制)至100 μL,制成接头工作液。引物23mer和19mer由上海基康生物技术公司合成,序列分别为23mer:5′-GATCCTGAGCTCGAATTCGACCC-3;19mer:5′-GGGTCGAATTCGAGCTCAG-3′。
1.2.41号染色体的体外扩增
在去蛋白的染色体DNA中加入2 μLSau3AI(0.01 U/μL,1×T4 DNA连接酶缓冲液配制),37 ℃酶切2 h,于70 ℃下20 min灭活Sau3AI酶。加入2 μL上述制备好的Sau3AI接头(50 ng/μL)、1.5 U T4 DNA连接酶(3 U/μL),16 ℃连接过夜(连接反应终体积为24.5 μL),最后于70 ℃下20 min灭活连接酶。经上述酶切、连接后的单染色体即可进行PCR扩增。PCR反应的总体积为60 μL,6 μL 10×Taq酶缓冲液、4 μL MgCl2(25 mmol/L)、8 μL dNTPs(2.5 mmol/L)、1 μL 19碱基引物(20 μmol/L)、0.5 μL TaqDNA聚合酶(5 U/μL),无菌ddH2O补至60 μL。用ABI Veriti 96 Well 型PCR仪进行扩增,扩增程序:94 ℃变性5 min,35个循环94 ℃ 1 min→50 ℃ 1 min→ 72 ℃ 1 min,最后72 ℃延伸15 min。第1轮PCR扩增在酶切、连接单染色体DNA的同一PCR管中进行。第2轮PCR扩增从第1轮PCR产物中取2 μL作为模板,其他反应成分不变,PCR反应程序与上述方法相同,将35个循环降至20个循环。此步骤设基因组DNA为模板的阳性对照和严格不含染色体DNA的阴性对照。
1.2.5尼罗罗非鱼基因组DNA的提取和纯化
取1 μg基因组 DNA用Sau3AI酶 37 ℃酶切过夜,与阴性对照、阳性对照和单染色体LA-PCR第2轮的扩增产物一起在1.0%琼脂糖凝胶低电压电泳直到完全跑开。进行 Southern 印迹转膜,以及地高辛(Digoxin)标记系统的非同位素杂交。用经EcoRI酶切、DIG 标记的尼罗罗非鱼基因组 DNA 作为探针进行 Southern blot。具体操作步骤参照罗氏地高辛标记DNA和检测试剂盒(DIG High Prime DNA Labeling and Detection Starter Kit I)说明书。
1.2.6微卫星检测
以1号染色体扩增产物为模板,基因组DNA为阳性对照,使用已在尼罗罗非鱼基因组上定位的两对微卫星引物UNH971和UNH106进行 PCR 检测(表1)。PCR 反应体系含1 μL模板、0.4 μL(10 mmol/L) dNTP、1 U Taq 酶、1.5 μL(5 mmol/L)引物、2 μL 10×Buffer,用无菌水补足20 μL。PCR反应程序为:95 ℃预变性 5 min、95 ℃变性30 s、58 ℃退火30 s、72 ℃ 延伸30 s,共35个循环:72 ℃ 延伸10 min。反应完毕后,1%的琼脂糖凝胶电泳检测扩增产物。
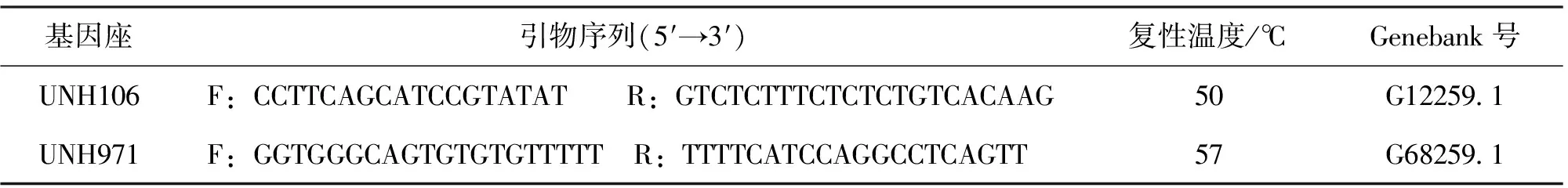
表1 两对微卫星引物特征Tab.1 Two pairs of microsatellite primers’ characteristics
1.2.7单染色体微克隆文库的构建
第二轮 LA-PCR 扩增产物采用天根DNA胶回收纯化试剂盒回收纯化,取1 μL与pGEM-T Easy Vector(Promega)载体连接,采用热击转化法将重组片段导入感受态细胞JM109中,并将转化菌涂于含有氨苄青霉素(Amp)、异丙基硫代半乳糖苷(IPTG)和 5-溴-4-氯-3-吲哚-β-D-半乳糖苷(X-gal)的LB平板上37 ℃培养过夜,计算 6个板的阳性克隆数。在超净工作台上挑取白斑菌于LB培养基中并加入15%甘油于-80 ℃下长期保存。从文库中随机选取部分重组克隆子并采用TaKaRa质粒提取试剂盒提取质粒。以19碱基引物对部分重组克隆子进行扩增,其中20 μL扩增体系:2.0 μL 10×Taq Buffer(含Mg2+)、4 μL dNTPs(2.5 mmol/L )、1 μL 19碱基引物(10 μ)、0.5 μL Taq DNA聚合酶(2.5 U/μL )、质粒0.5 μL和 ddH2O 1 μL;PCR 扩增程序:94 ℃ 预变性 5 min,94 ℃ 变性 1 min,58 ℃复性 45 s,72 ℃延伸 2.5 min,共 30 个循环,72 ℃总延伸 8 min。扩增结束后,1.0%琼脂糖凝胶电泳检测。
2 结果与分析
2.1 染色体的识别与标本制备
对尼罗罗非鱼60个分散清晰的细胞中期分裂相进行计数,如表2,其染色体数目主要是在39~44之间,其中80%的染色体数目为44。因此尼罗罗非鱼染色体数是2n=44。

表2 从60个细胞计算出的尼罗罗非鱼前中期染色体的数目Tab.2 Distribution of chromosome numbers in 60 metaphases cells from O.niloticus
2.2 染色体的显微切割
在切割过程中,激光器的能量越高,越易造成切割部位染色体的灼伤,激光器的能量过低,切割越困难。因此切割时需要依据不同部位调整能量,以确保可以切割下来又不会造成染色体灼伤。经过反复实验表明,采用莱卡公司LMD 6000激光显微切割系统,当激光输出值为20~40,速度为2~4,能取得较好的切割效果,样品损伤光斑直径可小于1 μ。图1为激光显微切割前后的染色体对比图,从图中可以看到分离了完整的一对1号染色体。

图1 尼罗罗非鱼中期染色体及1号染色体的分离Fig.1 The metaphase chromosome and chromosome 1 separation in O.niloticus
2.3 1号染色体的体外扩增
图2为第2轮扩增的结果, 二次分离的1号染色体扩增产物片段大小均为250~2 000 bp,尼罗罗非鱼基因组DNA为扩增模板的阳性对照所得产物片段,较1号染色体扩增产物的范围宽,阴性对照未出现扩增信号,说明没有外源DNA的污染。
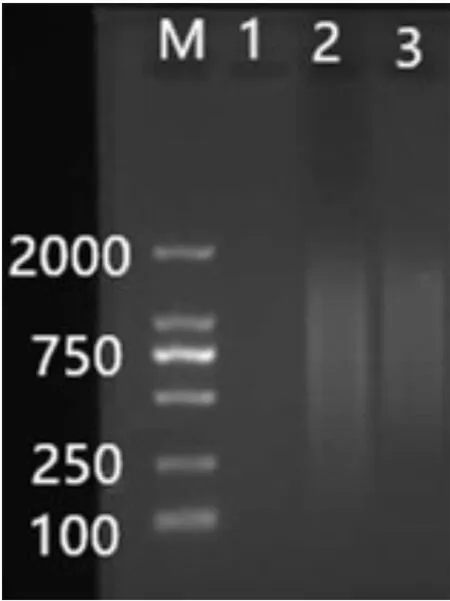
图2 LA-PCR第2轮扩增产物琼脂糖凝胶电泳图Fig.2 Image of agarose gel electrophoresis of the LA-PCR 2nd round amplification’s products
2.4 Southern blot验证
用经Sau3AI酶切、DIG 标记的尼罗罗非鱼基因组 DNA 作为探针对 LA-PCR 第二轮扩增产物进行Southern blot 验证,结果如图3。阳性对照、Sau3AI酶切的基因组DNA和单染色体扩增产物都有明显的杂交信号,而阴性对照则没有出现。结果表明,单染色体的扩增产物确实来自尼罗罗非鱼的基因组。
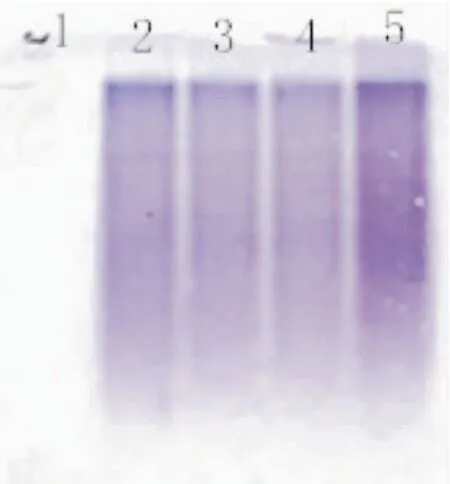
图3 Southern blot验证Fig.3 Verification with Southern blot
2.5 SSR检测
已定位在尼罗罗非鱼基因组上的 SSR 引物对扩增产物进行 PCR 分析,结果显示引物对UNH971,UNH106从1号染色体的扩增产物中能够扩增出目的条带(图4)。这表明单染色体扩增产物确实是来自尼罗罗非鱼基因组,本试验对尼罗罗非鱼1号染色体的分离和扩增是成功的。
图4 SSR引物对单染色体扩增产物的 PCR分析结果Fig.4 Results of PCR analysis on single chromosomes 1 amplification products of SSR primers
2.6 尼罗罗非鱼1号染色体质粒文库的构建立
尼罗罗非鱼1号染色体第 2 轮LA-PCR 扩增产物纯化回收后与 pGEM-Easy Vector连接,采用热击转化法将重组片段导入JM109感受态细胞并将转化菌涂于含有Amp、IPTG和 X-gal 的6个LB平板上37 ℃培养过夜,均长出密度适中的白色菌落,平均每板约有146个阳性克隆。19mer引物扩增部分重组质粒,1.0%凝胶电泳检测结果如图5显示,克隆片段长度分布在300~600 bp,平均为400 bp左右。
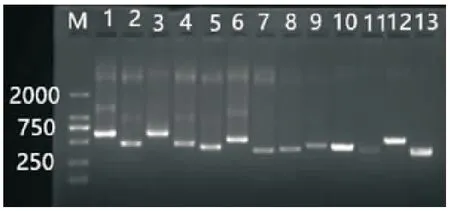
图5 部分重组质粒经19mer引物扩增后电泳结果Fig.5 Electrophoresis results of some recombinant plasmid with 19mer primer
3 讨论
3.1 尼罗罗非鱼染色体标本制备过程中的操作优化
鱼类染色体的研究已有半个多世纪,累积了多样化的染色体标本制备方法。本研究主要在原先的体腔注射PHA空气干燥法上做了细微调整[18],将原先7 h内的PHA作用时间延长至28~30 h,制得较理想的染色体标本。鱼体内注射适量的PHA可以起到增加细胞中期分裂相数目的作用[19],但因养殖温度、注射剂量和注射时间等多种因素的影响也会导致细胞分裂相数量不足或直接导致试验鱼死亡。经反复摸索发现罗非鱼水温在20~25 ℃时,按鱼体重以10 μg/g的量注射1 mg/mL的 PHA,暂养28~30 h后得到的处于分裂中期的头肾细胞较多。罗非鱼染色体较小,长度只有2~5 μm,需按鱼体重以2 μg/g的量注射0.2 mg/mL的秋水仙素,秋水仙素作用时间控制在1.5~2 h,不宜过长。作用时间过长,则易导致染色体收缩过度,呈点状,影响染色体的形态。在低渗中将时间延长至1 h,每隔10 min上下轻轻翻动离心管数下,使细胞充分悬浮并膨胀。卡诺氏固定剂中冰醋酸具有脱嘌呤作用[20],为避免染色体DNA受损,需把卡诺氏固定剂中的甲醇、冰乙酸的比例调整为4∶1,且每次固定时间缩短为15 min,有利于减小对染色体的伤害,保证染色体的完整性,并获得背景干净、染色体分散的标本。此外,膜片的制备与普通玻片不同,膜片不能使用酒精灯火焰加热。本团队摸索出滴片后,将膜片放在55 ℃烘箱中烘干10 s的方法,成功制备出高质量的有丝分裂中期染色体膜片。膜片材质极其轻薄,在操作时要小心谨慎,轻微的触碰都会使膜破裂。
3.2 单染色体显微分离技术优化
激光显微分离染色体和手工分离染色体是显微分离单条染色体的方法。手工分离染色体对操作者的熟练度具有较高的要求,同时容易出现机械接触,造成污染[21]。而染色体微分离、微克隆研究中应严格避免外源污染[22],本研究采用Leica LMD 6000正文激光显微切割系统,染色体滴片在POL类型的钢化薄膜上,用激光切割目标染色体,在重力作用下脱落进入收集管中。此操作不需借助外力,没有人为接触,避免了人为污染[23]。使用激光显微系统分离单染色体,对目标染色体进行可视化切割,具有更高的准确度,并能快速获得大量染色体或染色体片段。本团队研究发现目标染色体周围有其它染色体或细胞核成分时,可先通过微弱能量的激光束将干扰成分剔除 。若切割后发现被切下的承载目标染色体的膜粘附在载片膜的背面时,需适当增加激光能量输出值,增大激光光圈,并降低激光发射速度,使其直接脱落在下方备好的收集管中。尼罗罗非鱼的染色体属于中、小型染色体,要比大部分细菌还小,为避免外源DNA的污染,LA-PCR扩增等操作过程需在超净工作台内完成,并对所用试剂、水进行严格的无菌处理,严禁有任何的污染影响实验。
3.3 染色体文库的验证与分析
扩增产物经限制性内切酶Sau3AI酶切,并用地高辛(DIG-High Prime)标记尼罗罗非鱼基因组 DNA 为探针,对扩增产物进行Southern blot 杂交验证。本实验的Southern 杂交结果显示扩增产物来自尼罗罗非鱼基因组,且阴性对照没有出现信号,证明本实验没有被污染。此外,本研究应用第二代罗非鱼遗传连锁图上定位的微卫星做引物,并成功地在分离的1号染色体扩增产物上扩增出目的条带。微卫星标记具有高度特异性[24],进一步说明分离的1号染色体扩增产物来源的真实性。
本研究从尼罗罗非鱼头肾细胞入手,运用单染色体的微分离与微克隆技术,成功建立尼罗罗非鱼1号染色体的微克隆文库构建的方法,为该染色体的特异探针筛选及性别相关基因定位提供新的思路与方法,进一步为更好地开展尼罗罗非鱼分子辅助育种提供技术支撑。
