橡胶产品中多环芳烃提取前处理净化方法的验证
2018-07-09通标标准技术服务上海有限公司
/ 通标标准技术服务上海有限公司
0 引言
目前,固相萃取装置在食品、高分子材料等产品的色谱分析测试的前处理[1]中得到了广泛的应用[2-3],其具有简便、快速、高效等优势。常见的固相萃取装置有自制填料固相萃取柱和市售的固相萃取小柱。就自制填料固相萃取柱而言,填料的选择[4-5]和装填[6-7]手法及后续的洗脱溶剂的选择[8]等因素都直接影响其净化效果。本文通过对不同粒度的填充料的选择(硅胶)和合理的洗脱溶剂来验证本实验所选择的填充方式及洗脱方式的优越性。就市售固相萃取小柱而言,在实际使用中,洗脱溶剂的使用量和洗脱时间直接影响其净化效果。本文通过对橡胶产品中多环芳烃的测试,验证了本方法中前处理时所选择的洗脱溶剂和洗脱时间的优越性,有效地将目标物与干扰组分相分离,大大增强对目标物特别是痕量目标物的检出能力,提高了被测样品的回收率[9-10]。
1 实验方法
1.1 前处理步骤
准确称取制样为 5 mm×5 mm×5 mm 的橡胶样品1g,使用(正己烷∶二氯甲烷=1∶1)的溶剂超声提取多环芳烃类,超声提取液旋蒸浓缩至约1 mL,经固相萃取小柱净化后,使用GCMS对多环芳烃类物质进行分析定量。
1.2 仪器及分析参数
Agilent 7890A(GC)-5975C(MS):
1)色谱柱:DB-5MS,柱长为30 m,内径为0.25 mm,膜厚 0.25 μm(J&W);
2)柱温度程序:初始温度60 ℃,以30 ℃/min升温至120 ℃,再以10 ℃/min升温至250 ℃,再以10 ℃ /min 升温至 310 ℃,保持 2.5 min;
3)进样口温度:250 ℃;
4)色谱-质谱接口温度:280 ℃;
5)离子源温度:230 ℃;
6)载气:氦气,纯度≥99.999%,1.0 mL/min;
7)电离方式:EI;
8)电离能量:70 eV;
9)质量扫描范围:50~450 amu;
10)测定方式:选择离子监测方式;
11)进样方式:不分流进样,2.0 min后开阀;
12)进样量:1.0 μL;
13)溶剂延迟:3 min。
1.3 实验试剂和耗材
正己烷:色谱级;二氯甲烷:色谱级;石油醚:95% 石油醚(沸程40~60 ℃),纯度95%;层析硅胶:60~100目、200~300目;40 mL螺纹管;旋转蒸发仪;玻璃层析柱:直径×高度= 1.5cm×15cm;乙二胺-N-丙基填料固相萃取小柱:2g/6 mL;固相萃取装置。
1.4 标准品
18种多环芳烃标准品:纯度≥96%,分别为:萘、苊、苊烯、芴、蒽、菲、荧蒽、芘、苯并(a)蒽、苯并菲(屈)、苯并(b)荧蒽、苯并(k)荧蒽、苯并(j)荧蒽、苯并(e)芘、苯并(a)芘、茚苯(1,2,3-cd)、二苯并(a,h)蒽、苯并(g,h,i)苝(安谱)。1.4.1 标准储备溶液(约 1 000 mg/L):准确称取适量18种多环芳烃标准品,用正己烷将每种物质配成浓度约为1 000 mg/L的标准储备液。
1.4.2 混合标准工作溶液(10 mg/L):准确移取适量标准储备溶液(1.4.1),用正己烷配制成浓度为10 mg/L的混合标准工作溶液。
1.4.3 工作曲线配制:将混合标准工作溶液移取适当体积至容量瓶中,分别配制成浓度为0.5 mg/L、1 mg/L、2 mg/L、5 mg/L、10 mg/L 的 5 个浓度点。

表1 化合物的曲线方程及相关系数
1.5 结果计算
样品中多环芳烃含量按式(1)计算,结果表示到小数点后一位。计算结果需将空白值扣除。

式中:Xi—— 样品中多环芳烃i的含量,mg/kg;
ci—— 标准工作溶液中多环烃i的浓度,mg/L;
Ai—— 样液中多环芳烃i峰面积;
As—— 标准工作溶液中多环芳烃i峰面积;
V—— 样液最终定容体积,mL;
F—— 稀释因子;
m—— 最终样液代表的试样量,g
2 自制硅胶填料净化柱填装验证
2.1 硅胶的选择和失活处理
进行硅胶选择时,颗粒很细的硅胶净化效果好,但样品净化时间长,对于基质粘稠样品所需净化时间更长,这样处理样品数量多时实验室效率低;颗粒较大的硅胶不能起到很好的净化效果,所以本文验证了60~100目和200~300目层析硅胶结合使用的最佳选择量。
硅胶在装填柱子前需要进行失活处理,将层析用硅胶在130 ℃的烘箱中烘12 h,置于干燥器中冷却。装填柱子时,在装有硅胶的烧杯中加入10%(质量比)的去离子水,搅拌均匀,使其占居部分硅胶的位点,避免净化过程中因硅胶吸附太强而不能连续洗脱出的现象发生。另外,部分溶剂与硅胶作用产生热,容易使硅胶柱出现气泡(如二氯甲烷做溶剂),需要在装填柱子时,再使用该洗脱溶剂进行失活处理(方法:加入一倍于硅胶体积的溶剂,搅拌均匀,本文使用该方法对层析硅胶进行了失活处理),然后装填。
2.2 装填
准备干净的层析柱,用玻璃棉(0.5~1cm高)拦截,玻璃棉上面用无水硫酸钠填充(至1cm高),称取10g使用水去活的层析硅胶(选择60~100目∶200~300目=7∶3,验证数据见表2),填充无水硫酸钠上层,晃动玻璃层析柱,不断用滴管乳胶头敲实,待柱子顶部平整不再有凹陷,在硅胶层的顶部加入1cm的无水硫酸钠层,继续敲实。依次加入20 mL的石油醚,30 mL的正己烷平衡硅胶柱。柱子不能干涸。
2.3 洗脱溶剂的选择
依据有机溶剂极性,选择溶解性和亲和性适合多环芳烃类化合物,且更易洗脱出多环芳烃类的溶剂进行洗脱,依据表3数据,使用100 mL的(正己烷∶二氯甲烷=7∶3)混合溶液淋洗。
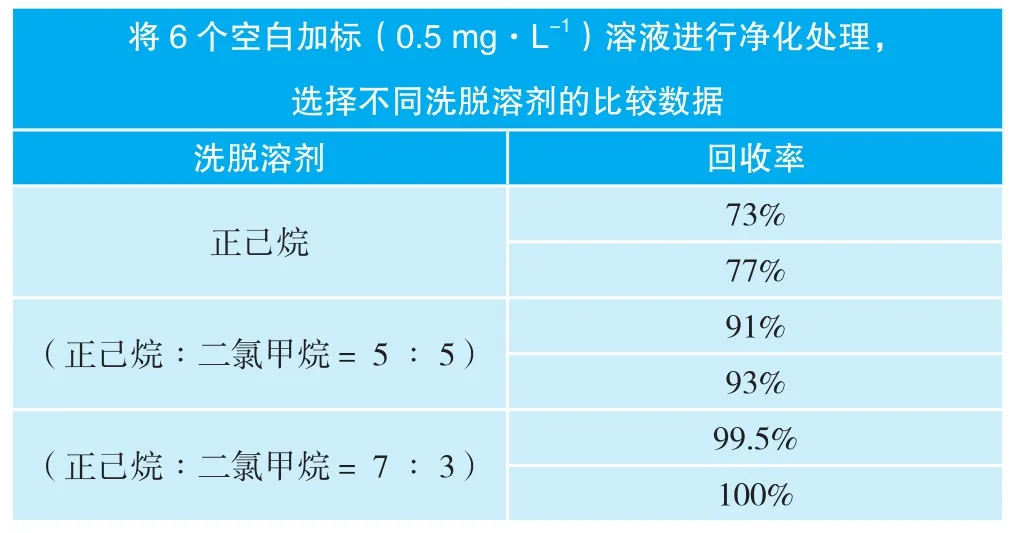
表3 洗脱溶剂选择比较
2.4 上样和淋洗液的收集浓缩
收集淋洗溶液至250 mL的平底烧瓶中,在50 ℃,300 bar的条件下将淋洗液旋转蒸发至小于3 mL,使用正己烷定容至10 mL,供GC-MS分析。
3 乙二胺-N-丙基填料固相萃取小柱淋洗曲线验证
3.1 乙二胺-N-丙基填料固相萃取小柱净化流程步骤
针对乙二胺-N-丙基填料固相萃取小柱的洗脱,选取第三方检测中检出次数最多的化合物萘,做空白加标,对洗脱效果做了淋洗曲线验证。
依据验证结果,确证乙二胺-N-丙基填料固相萃取小柱净化流程步骤见图1。乙二胺-N-丙基填料固相萃取小柱淋洗曲线数据见表4。
4 两种固相萃取小柱净化数据
4.1 自制固相萃取小柱净化结果
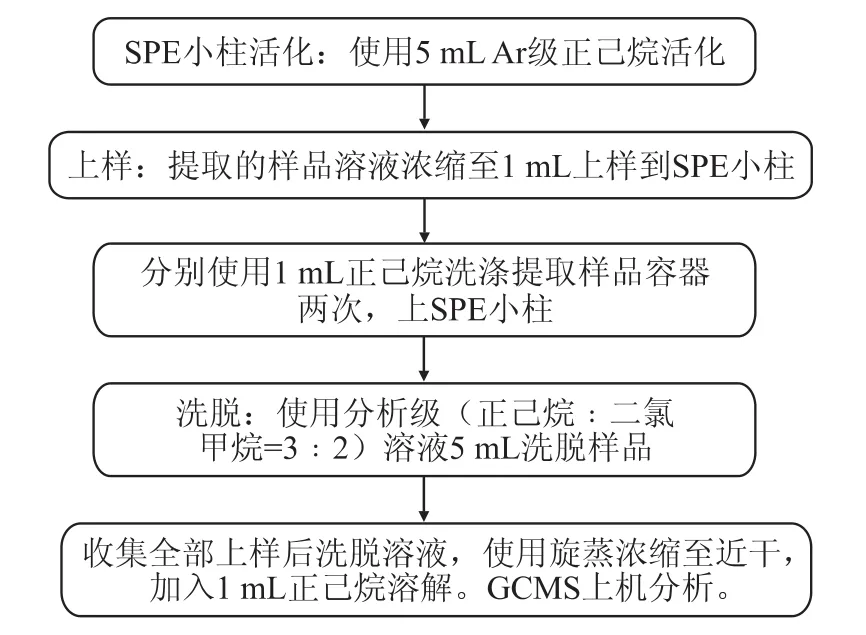
图1 净化流程-乙二胺-N-丙基填料固相萃取小柱
使用自己填装的层析硅胶固相小柱,对未检出橡胶基质样品加标(加标理论浓度0.5 mg/kg)前处理溶液进行净化、分析、计算结果回收率(表5),回收率范围在80%~130%之间;通过标准偏差评价测试结果的重复性(表6),8个基质加标样品,18种多环芳烃类物质的标准偏差<5%。

表4 乙二胺-N-丙基填料固相萃取小柱淋洗曲线数据表
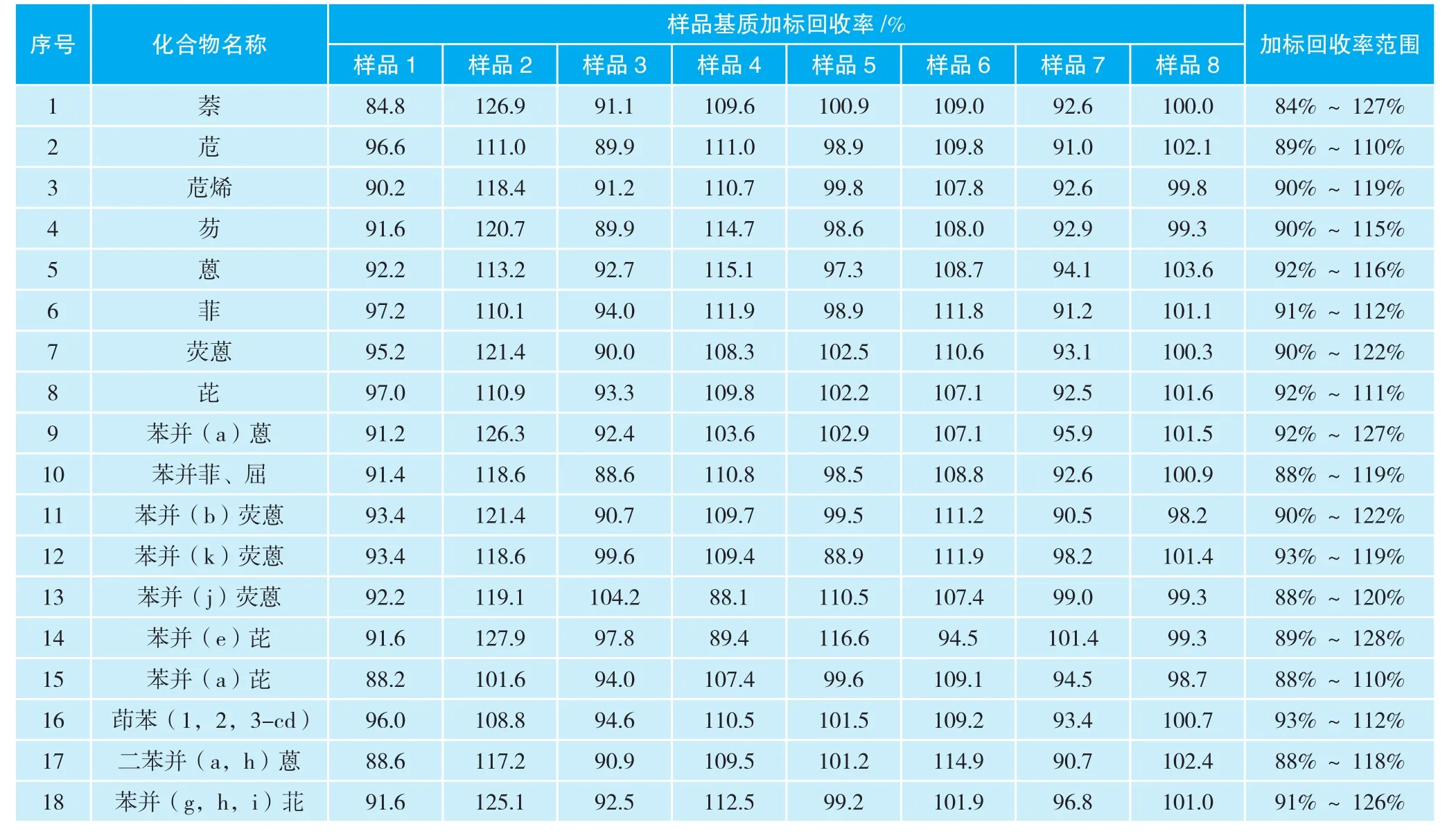
表5 样品基质加标回收率数据

表6 样品基质加标重现性数据
4.2 市售固相萃取小柱净化结果
做市售乙二胺-N-丙基填料固相萃取小柱空白验证,验证结果未检出。使用市售乙二胺-N-丙基填料固相萃取小柱,对橡胶基质样品加标(加标理论浓度0.1 mg/kg)前处理溶液进行净化、分析、计算结果回收率(表7),18种多环芳烃类物质回收率范围在85%~95%。

表7 样品空白和加标回收率数据
4.3 有值样品使用和不使用固相萃取小柱净化数据比较
通过检测有值橡胶产品,并对数值比较,验证检测结果的重复性,两种净化方式标准偏差<5%,详见表8、表9。

表8 层析硅胶柱有值样品数据比较

表9 乙二胺-N-丙基填料柱有值样品数据比较
4.4 讨论
通过上述实验,证明了使用本文选择的两款净化小柱进行橡胶类产品的前处理的实验结果准确可靠,重现性好。实验中利用层析硅胶柱从非极性体系中吸附极性化合物的性质,使得极性物质被吸附于硅胶上。利用相似相溶原理,使用弱极性溶剂进行洗脱,样品中的多环芳烃类物质会被洗脱流出,实现对样品的纯化。在实验进行仪器分析时,对于基质复杂的样品,色谱图上会出现很大的背景峰(见图2),使用层析硅胶柱净化后,在色谱图上背景峰被去除(见图3)。同样,应用乙二胺-N-丙基小柱正相作用原理,对极性化合物进行吸附,使杂质直接流出,实现对样品的纯化。
纯化后的样品溶液,GCMS分析干扰少,定量更准确,效率更高。且得黏稠提取溶液不用大体积稀释可被GCMS检测,检测限低;净化后的溶液使用GCMS分析,使仪器部件耗材使用更长的时间,节约仪器耗费成本。
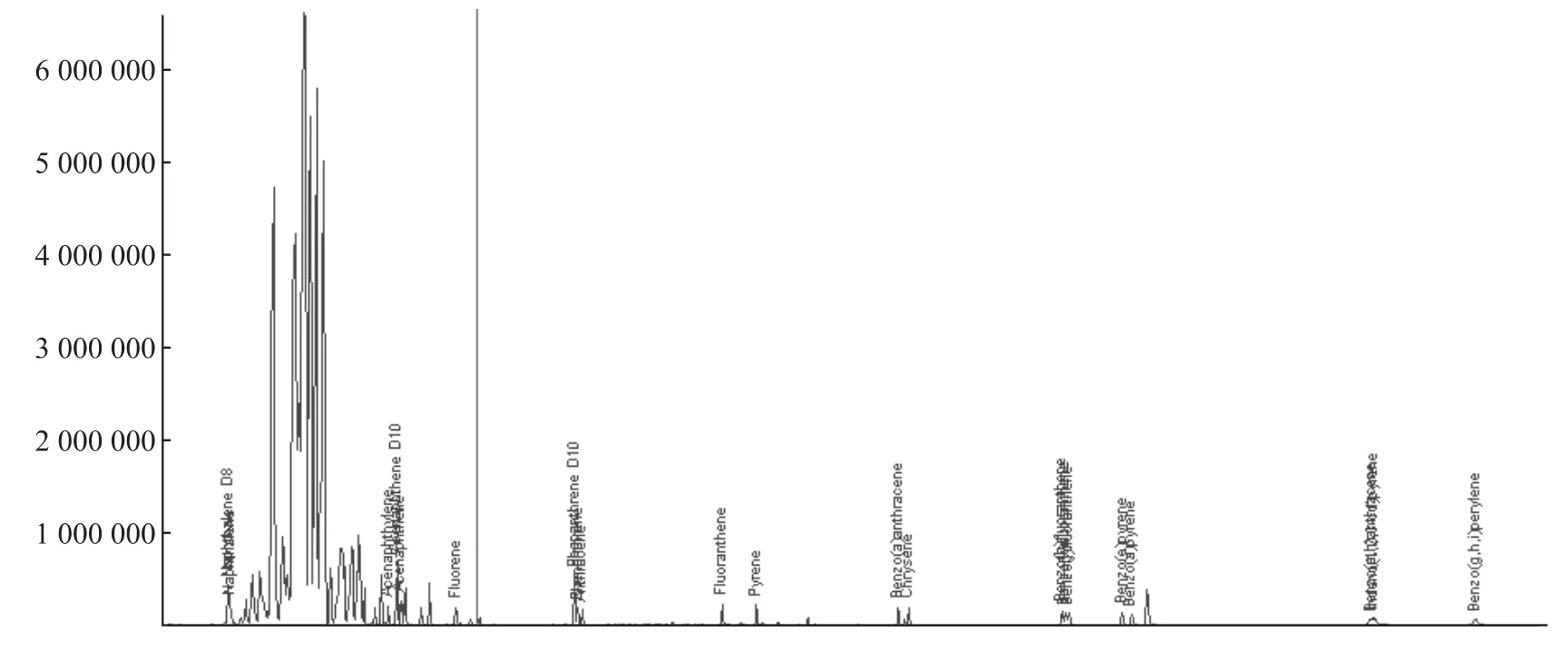
图2 未使用层析硅胶柱净化谱图
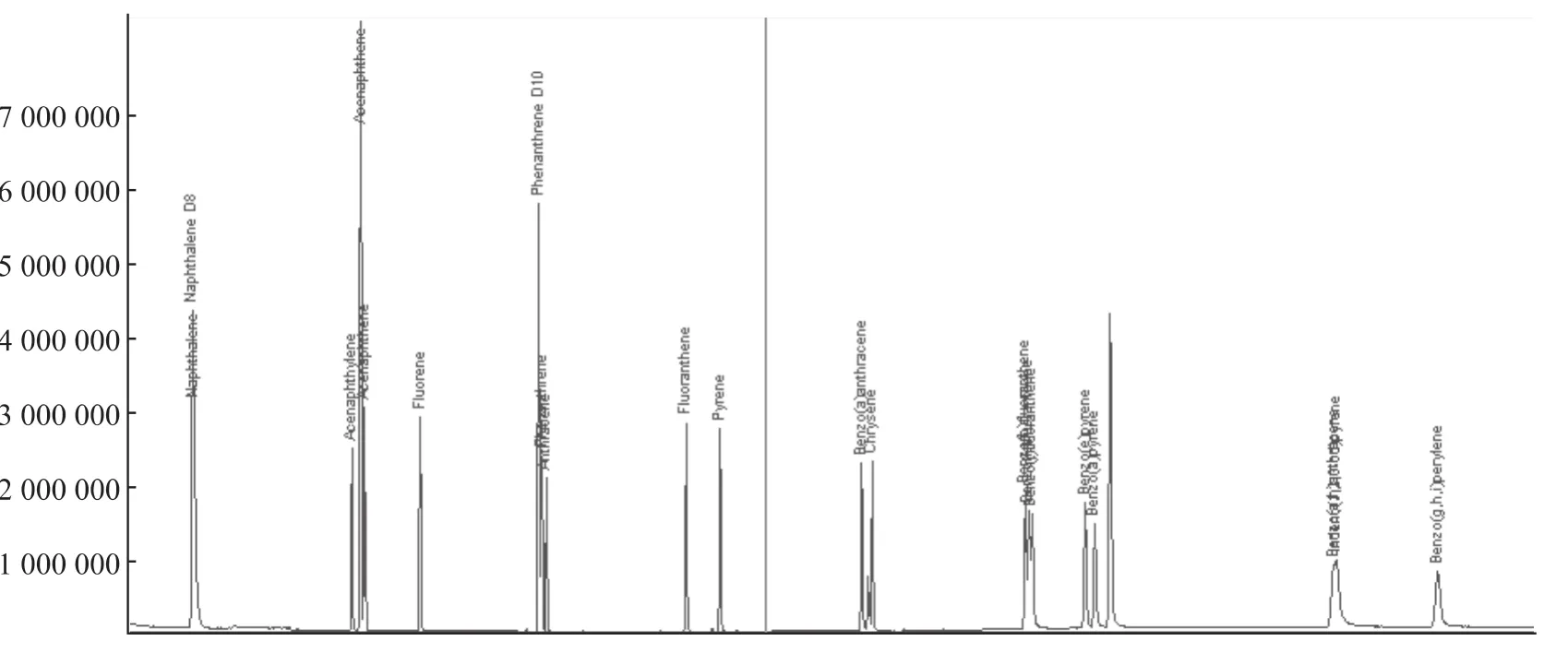
图3 使用层析硅胶柱净化谱图
5 结语
对橡胶产品中多环芳烃提取前处理溶液的净化,自制硅胶填料固相萃取小柱和市售乙二胺-N-丙基填料固相萃取小柱对于有基质干扰的样品,能够很好地去除基质干扰,确保分析检测结果准确可靠;其中,自制硅胶填料固相萃取小柱实验成本低,可以根据样品基质黏稠程度,填制大的硅胶柱,接受大的上样量;市售乙二胺-N-丙基填料固相萃取小柱实验流程时间短,效率高。但是,自制硅胶填料固相萃取小柱需要自己制备,实验流程时间长;乙二胺-N-丙基填料固相萃取小柱一般市售填料量少,不能接受大的上样量,如果根据需要购买填料量大的这种柱子,则价格高。
实验中,实验人员需要根据具体实验要求的完成时间和样品黏稠程度选择合适的固相萃取小柱。两种净化方式,针对经济型,一般选择自填固相萃取小柱;针对节约时间成本,选择市售固相萃取小柱。
[1]马建明,龚文杰.柱层析分离净化的实验方法和技巧探讨[J].中国卫生检验杂志,2008,18(4):745+762.
[2]宋冠群,林金明.环境样品中多环芳烃的前处理技术[J],环境科学学报,25(10):1287-1296.
[3]覃素姿,王宛,王茹意.固相萃取法和GC-MS联用检测石油污染物中17种多环芳烃[J].广东化工,2014,41(11):227-229.
[4]贾瑞宝,孙韶华,刘德珍.用固相萃取技术富集水中多环芳烃[J].色谱,1997(11):524-526.
[5]胡健,张国平,刘丛强.固相萃取柱净化-液相色谱法测定大气中多环芳烃[J].地球与环境,2014(Z1):94-97.
[6]卢福峰,邢核,许秀艳,等.ASE萃取-SPE净化-HPLC法测定土壤中多环芳烃[J].环境监测管理与技术,2007(3):25-27.
[7]郑慧,齐邦峰,程仲芊,等.HPLC法测定橡胶填充油中稠环芳烃含量的研究[J].化工科技,2011(2):34-37.
[8]傅若农.近年国内固相萃取-色谱分析的进展[J].分析试验室,2007(2):100-122.
[9]董新昕.欧盟制定多环芳烃使用限量[N].中国国门时报,2004-09-06.
[10]周翔.快速溶剂萃取-SPE硅胶柱净化-高效液相色谱法测定土壤中多环芳烃16种化合物的研究[J].环境研究与监测,2010(5):57-58.
