丙酮酸脱氢酶复合物缺乏症1例
2018-07-06郎继荣韩凝
郎继荣 韩凝
丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHc)是糖有氧氧化的限速酶,其不可逆的催化由糖酵解反应生成的丙酮酸为乙酰CoA,后乙酰CoA参与到三羧酸循环中氧化磷酸化产生能量。PDHc缺乏症是一种较罕见的遗传代谢性疾病,由于基因突变造成丙酮酸代谢障碍,导致乳酸堆积和能量生成障碍,临床常表现为乳酸酸中毒,神经发育迟滞,肌张力低下等,由于其发病率低,国内病例报道少,临床表现复杂,容易造成漏诊及误诊。本文报道一例PDHc缺乏症患者,并复习相关文献,对其临床特点进行探讨,以提高临床医生对本病的认识。
1 临床资料
1.1 发病情况患儿,男,12岁,主因双下肢无力10年余,加重3 d入院。患者于10余年前感冒、发热、腹泻后出现双下肢活动不灵,双上肢基本正常,就诊于当地儿童医院,行肌电图检查,提示肌源性损伤(自诉),口服维生素类营养药物后好转,日常活动不受限。后患者因体育成绩较差,呈运动不耐受,如跑步后数分钟,需休息几分钟方能继续跑,于2015年8月就诊当地市级医院,行头颅MRI、MRI强化及肌电图检查,具体诊断不详,给予中药治疗,效果不佳,仍运动不耐受。患者于3 d前军训、感冒后再次出现四肢无力,表现为行走需要帮助,上肢抬起困难,症状逐渐加重,于2017年7月就诊于河南宏力医院。无明确既往史;患者父亲(Ⅰ2)、母亲(Ⅰ1)及弟弟(Ⅱ2)既往体健(图 1)。
1.2 体格检查血压:90/60 mmHg,脉搏正常,心脏、肺部听诊无异常,神志清楚,反应正常,精神差,言语音量小,双眼球活动充分,双眼睑闭合有力,闭唇有力,面部感觉正常,余颅神经查体无异常。抬头、耸肩力弱3级,双上肢近端肌力2级,远端4+级,双下肢近端3级,远端4+级,四肢腱反射均减低,双侧病理征阴性。
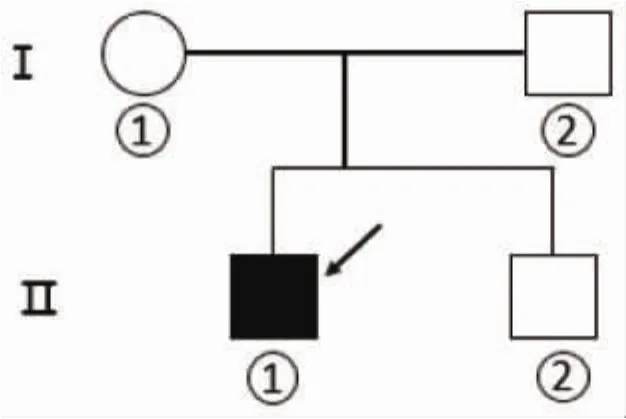
图1 家系图
1.3 辅助检查生化全项 (2015年 8月):丙酮酸增高:190.5 mmol/L(20~100 mmol/L)和乳酸增高:5.67 mmol/L(0.7~2.1 mmol/L)。 MRI及 MRI强化(2015年 08月):双侧苍白球对称性病变,强化未见明显异常 (图2A~C);肌电图(2015年8月):四肢广泛肌源性损伤;头颅MRI+DWI(2017年9月):双侧苍白球病灶较前明显增大(图3)。抽取患者、父母、弟弟、小姨和舅舅外周静脉血后送检,基因学检查显示患儿PDHA1基因编码区第376号核苷酸由C变为 T(c.376C>T),导致第 126号氨基酸由 Arg变为 Cys(p.Arg126Cys);其母为杂合子(见图 4A、B)。
1.4 诊断、治疗及随访入院后依据患者临床表现及辅助检查结果考虑为PDHc缺乏症,给予左旋肉碱、精氨酸、辅酶Q10和维生素B1等治疗。后患者因呼吸系统感染转至当地省级医院,于2017年9月随访发现,患者目前仍存在四肢无力,双下肢不能站立,左上肢可抓东西。头颅MRI+DWI(2017年09月):双侧苍白球病灶较前明显增大(图3A~E)。
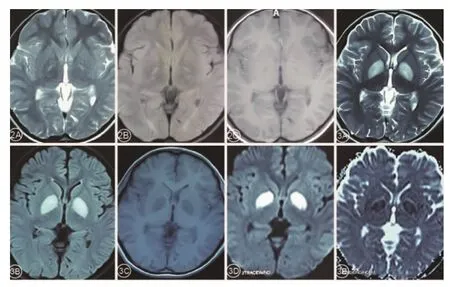
图2 头颅MRI(2015年8月) 2A-2C分别为 T2加权像、FLAIR像、T1增强像;2A、2B显示双侧苍白球对称性高信号,2C显示病灶未见明显强化图3 头颅MRI(2017年9月) 图3A-3E分别为T2加权像、FLAIR 像、T1增强像、DWI和 ADC;3A、3B、3D 显示双侧苍白球高信号,较第一次MRI病灶范围明显增大,3C未见明显强化,3E显示双侧苍白球低信号,提示为细胞毒性水肿

图4 患儿与患儿母亲测序结果 图4A:患儿样本序列chrx:193681199位置有c.376C>T的纯合子突变;图4B:患儿母亲样本序列chrx:193681199位置有c.376C>T的杂合子突变
2 讨论
丙酮酸脱氢酶复合物缺乏症在1970年由BLASS等人[1]首先报道,其主要表现为高乳酸血症、进行性神经和肌肉变性,常于童年死亡。此患者主要表现为运动不耐受和高乳酸血症,并且MRI显示双侧苍白球异常信号,此表现与leigh病典型影像学表现基本一致,并进一步行基因学检查,最终诊断为PDHc缺乏症。PDHc是由3种酶组成的复合体,其中丙酮酸脱氢酶是由两个α和β亚基组成的四聚体,其分别由PDHA1和PDHB编码。PDHA1基因是E1α缺乏最常见的突变基因,其位于X染色体的Xp22.1-22.2上,可有多个突变点,表现为X连锁隐性遗传。此患者基因学检测发现患儿及其母亲的PDHA1基因相同位置的突变点,符合X连锁隐性遗传的特点,其母亲为基因携带者(杂合子)。患者为男性,仅有一条X染色体,为纯合子,故产生临床症状。另外,患儿及其目前的突变点为第376个核苷酸由胞嘧啶突变为胸腺嘧啶,即c.376C>T,导致第126号氨基酸由 Arg变为 Cys(p.Arg126Cys),此突变在国内外尚未有报道。
丙酮酸复合物缺乏症的临床表现多样,与年龄、性别等均有关系。一项入组371例丙酮酸复合物缺乏症患者的研究发现[2],神经发育延迟和肌张力低下是其最常见的表现;脑结构异常主要表现为脑室扩大、胼胝体发育不良;年龄较小的死亡患者较存活患者相比,临床症状出现较早,血乳酸水平较高。PDHA1基因编码的丙酮酸脱氢酶E1α突变约占丙酮酸复合物缺乏症的70%,男性可表现为3种形式[3]:①表现为新生儿高乳酸血症伴大脑发育不良(如胼胝体发育不全);②常于婴儿和童年发病,主要表现为leigh病和间歇性共济失调;起病前5年表现为呼吸障碍、间断下肢无力和共济失调以及由于周围神经病导致的腱反射减弱,后主要表现为中重度的发育迟缓,一般最终发展为leigh病;③此型表现较少见,起病症状较轻,进食富含碳水化合物的食物后出现发作性共济失调,缓慢发展为leigh病,一些患者在婴儿期发展为急性周围神经病[4],有些发展为急性发作性共济失调且不伴有认知障碍[5]。女性患者的临床表现较一致,大部分伴有癫痫,一般出现于出生后的前6个月,常常被诊断婴幼儿痉挛或严重肌阵挛,并伴有先天畸形(小头畸形伴前额突出、朝天鼻、大鼻孔、宽鼻梁、长人中以及低位耳、短手指、四肢近端短小、断掌等)、中重度精神发育迟滞、四肢痉挛性瘫痪和脑瘫等。本例患者为男性,符合男性表现的第2型。
一般认为此病的预后较差,有研究[2]显示PDHA1基因位于378号氨基酸突变时,患者预后较差,甚至在出生后的2年就会死亡;其他2个常见氨基酸位点263和302号突变能够缩短患者寿命。此病缺乏规范的治疗方案,大部分病例报道都是给予维生素B1和生酮饮食,但剂量多种多样。有研究[6]显示生酮饮食能够缓解患者症状,减缓脑损害,长期的预后与规律的检测血清酮体水平和调整饮食搭配高度相关。国内吴莫龄等人[7]报道了1例丙酮酸脱氢酶复合物缺乏症患者,给予维生素B1、左旋肉碱、辅酶 Q10等治疗后,随访3年,患者症状好转,且复查头颅 MRI,病灶较前未扩大。本例患者2年内,症状及颅内病灶较前明显扩大,可能和不规律服用药物相关。临床上早期识别本病,早期给予生酮饮食、规律检测血清酮体,调整饮食搭配等治疗能够缓解患者症状、延长患者寿命。
[1]BLASS JP,AVIGAN J,UHLENDORF BW.A defect in pyruvate decarboxylase in a child with an intermittent movement disorder[J].J Clin Invest,1970,49(3):423-432.
[2]PATEL KP,O'BRIEN TW,SUBRAMONY SH,et al.The spectrum of pyruvate dehydrogenase complex deficiency:clinical,biochemical and genetic features in 371 patients[J].Mol Genet Metab,2012,105(1):34-43.
[3]DE MEIRLEIR L.Disorders of pyruvate metabolism [J].Handb Clin Neurol,2013,113:1667-1673.
[4]STRASSBURG HM,KOCH J,MAYR J,et al.Acute flaccid paralysis as initial symptom in 4 patients with novel E1 alpha mutations of the pyruvate dehydrogenase complex[J].Neuropediatrics,2006,37(3):137-141.
[5]DEBRAY FG,LAMBERT M,GAGNE R,et al.Pyruvate dehydrogenase deficiency presenting as intermittent isolated acute ataxia[J].Neuropediatrics,2008,39(1):20-23.
[6]SOFOU K,DAHLIN M,HALLBOOK T,et al.Ketogenic diet in pyruvate dehydrogenase complex deficiency:short-and longterm outcomes[J].J Inherit Metab Dis,2017,40(2):237-245.
[7]吴莫龄,刘丽,蔡燕娜,等.丙酮酸脱氢酶复合物缺乏症一例的临床特点及基因检测 [J].中华儿科杂志.2014,11(52):863-866.
