玉米秸秆不同发酵时期理化性状和细菌群落多样性
2018-07-06王秀红李欣欣史向远王保平杜慧平籍增顺
王秀红,李欣欣,史向远,周 静,王保平,杜慧平,籍增顺
(山西省农业科学院 现代农业研究中心,山西 太原 030031)
我国年产玉米秸秆约2.5亿t,用作秸秆还田和牲畜粗饲料的总量不足60%,其余均被焚烧,造成资源浪费和环境污染[1]。玉米秸秆是一种很好的可再生生物质资源,将秸秆通过好氧堆肥处理用作食用菌栽培料、设施园艺栽培基质以及有机肥具有很好的应用前景[2-4]。微生物是堆肥过程中发挥分解腐熟作用的关键因子,了解发酵过程中微生物的菌群变化不仅可以从菌群消长变化的角度为好氧发酵机制积累资料,而且能为改进玉米秸秆好氧发酵堆肥的工艺参数提供参考。通过传统平板培养仅能获得1%~15%的可培养微生物,无法反映堆体中微生物群落的真实情况[5]。随着分子生物学技术的发展,宏基因组学(Metagenomics)已成为当前研究环境微生物种群动态的有力工具,高通量测序技术可以避开传统可培养方法的局限性,能较全面地对生境中的微生物总DNA进行分析,灵敏、快速地检测环境样品中微生物群落结构多样性,从而获得复杂生境中微生物种群变化的准确信息[6-8]。利用宏基因组学技术可以深入挖掘堆肥生境中各种类型的降解菌,发现新菌种[9-10]。Martins等[11]首次利用高通量测序技术对堆肥生境的微生物总DNA进行研究,开启了对堆肥生境中高效降解菌群及其基因资源的认识,通过对圣保罗热带公园堆肥样品总DNA测序,获得300万条序列,并发现变形菌门的乳酸杆菌在该堆肥生境中占据优势;此外,通过功能基因预测的112个纤维素酶相关蛋白质中,有32个具有纤维素结合结构域;可见,宏基因组技术在堆肥生境中分析微生物群落结构变化上具有明显优势。De Gannes等[12]利用454焦磷酸测序分析了3种木质纤维素堆肥不同阶段的菌群,结果显示,随着堆肥进程的变化,微生物群落发生明显更替,堆肥嗜温期和腐熟期微生物群落存在差异,嗜温期微生物在腐熟期并不是先前推测的处于休眠状态,认为堆肥所处发酵阶段对微生物群落的影响比堆肥底物的类型更大。沈其荣等[13]研究表明,堆肥的有机质分解主要发生在建堆后的30 d之内,因此,掌握这一时期堆料好氧发酵过程中的理化性状及微生物的种类和丰度对于改进堆肥工艺、提高腐熟效果至关重要。堆肥过程中细菌是最主要的优势菌群[14],能够影响堆料的腐熟进程。但是,关于玉米秸秆好氧发酵不同翻堆时期的细菌群落演替情况在宏基因组水平上报道较少。
本研究通过高通量Illumina MiseqTM宏基因组16S rRNA测序,对玉米秸秆条垛式堆肥不同翻堆时期的细菌菌群进行分析,以期从不同物种分类角度掌握好氧发酵的微生物菌群演替规律,认识影响玉米秸秆好氧发酵的关键菌群,为筛选功能菌株、制备微生物复合菌剂以及加快发酵进程指明研究方向并提供理论支持。
1 材料和方法
1.1 试验设计
试验地点设在山西省农业科学院现代农业研究中心榆次东阳试验基地。玉米秸秆好氧发酵采用3.00 m×1.50 m×1.35 m 的条垛式堆肥方式进行。主要原料为玉米秸秆,主要辅料为干牛粪,2种物料分别来自东阳基地试验田和周边养殖户。将收集到的玉米秸秆粉碎至3~5 cm,干牛粪碾碎,每个条垛堆均为玉米秸秆粉350 kg和牛粪100 kg的混合,配以少量的生石膏粉、过磷酸钙、尿素和豆饼,将C/N调节至30左右,调节水分含量至65%~70%,物料均匀混合,共设3次重复。建堆后每7 d翻堆一次,共翻堆5次。建堆初期及5次翻堆均从堆体不同长度的纵切面即外层(1~10 cm)、中层(25~45 cm)和内层(60~75 cm)多点采样,均匀混合后分别标记为CS0、CS1、CS2、CS3、CS4和CS5(CS:Corn straw)共6个样品,用冰盒带回实验室。部分样品冷藏或自然风干用于理化指标测定;部分样品保存于-80 ℃超低温冰箱,送上海生工生物工程股份有限公司进行16S rRNA宏基因组测序和生物信息学分析。
1.2 试验方法
1.2.1 理化数据测定 理化指标测定参考刘雯雯[15]的方法进行。采用RC-4HC型温湿度记录仪实时测定距堆体外侧45 cm深度处温度;干燥失重法测定含水量;新鲜堆肥样品与去离子水按1∶10(m/V)混合,于水平摇床上振荡24 h,4 ℃ 12 000 r/min离心10 min,上清经单层滤纸过滤后收集,再用0.45 μm 微孔滤膜过滤得滤液,分别用pH计测定pH值、分光光度计测E4/E6值、TOC仪测定水溶性有机碳w(C)(Water-soluble organic carbon)和水溶性氮w(N)(Water-soluble nitrogen);风干样用TOC仪测定总有机碳、元素分析仪测定全氮、马弗炉高温灼烧测定有机物含量。
1.2.2 样品基因组DNA提取 参照OMEGA试剂盒E.Z.N.ATMMag-Bind Soil DNA Kit的试剂盒方法提取细菌基因组DNA,操作步骤参照试剂盒使用说明书。
1.2.3 Illumina MiseqTM宏基因组测序 PCR所用的引物已经融合了Miseq测序平台的V3~V4通用引物。341F引物:5′-CCTACGGGNGGCWGCAG-3′;805R引物:5′-AGACTACHVGGGTATCTAATCC-3′。30 μL PCR反应体系:2×Taqmaster Mix 15 μL,Bar-PCR primer F(10 μmol/L)1 μL,Primer R(10 μmol/L)1 μL,Genomic DNA 10~20 ng,加无菌水定容至30 μL。PCR反应条件:94 ℃3 min;94 ℃ 30 s,45 ℃ 20 s,65 ℃ 30 s,共5个循环;94 ℃20 s,55 ℃20 s,72 ℃ 30 s,共20个循环;72 ℃ 5 min。第1轮PCR结束后,引入Illumina桥式PCR兼容引物再进行第2轮扩增,扩增体系同第1轮,反应条件为:95 ℃30 s;95 ℃15 s,55 ℃ 15 s,72 ℃30 s,共5个循环;72 ℃5 min。PCR产物进行琼脂糖电泳检测,并对PCR产物DNA进行纯化回收(0.6倍的磁珠Agencourt AMPure XP处理),利用Qubit 2.0 DNA检测试剂盒对回收的DNA精确定量,样品等比例混合后上机测序。
1.3 序列分析
1.3.1 序列预处理 去除引物接头序列,再根据PE reads之间的overlap关系,将成对的reads拼接成一条序列,然后按照barcode标签序列识别并区分样品得到各样本数据,最后进行质控过滤,得到各样本有效数据。
1.3.2 操作分类单元(Operational taxonomic units,OTU) 利用Usearch软件将所有样本按照序列间的距离进行聚类,根据序列之间的相似性将序列分成不同的OTU。在97%的相似水平下的OTU进行生物信息统计分析。在OTU聚类结果的基础上,选择丰度最高的序列作为OTU的代表性序列,进行各类的OTU分析,从而了解样品测序结果中的菌种、菌属等信息。
1.3.3 OTU多样性分析 利用mothur软件计算堆肥样品细菌Alpha多样性指数。用chao1指数估计群落中含OTU数目;用Shannon指数和Simpson指数估算样品中微生物多样性,Shannon值越大,说明群落多样性越高,相反,Simpson指数值越大,说明群落多样性越低;Coverage表示各样品文库的覆盖率,其数值越高,样本中序列没有被测出的概率越低,实际反映了本次测序结果是否代表样本的真实情况。各指数相关计算公式如下。
chao1指数:
①
Shannon指数:
②
Simpson指数:
③
Coverage计算:
④
式中,Schao1为估计的OTU数目;Sobs为实际观测到的OTU数目;n1为只含有一条序列的OTU数目;n2为含有2条序列的OTU数目;ni为含有i条序列的OTU数目;N为序列总数。
1.3.4 物种分类 利用基于Bergey′s taxonomy的RDP classifier分类方式对OTU进行物种分类。
1.4 数据分析
采用Microsoft Excel 2007进行数据整理,以SPSS 21.0统计分析软件进行数据分析,采用Duncan多重比较法进行差异显著性分析。
2 结果与分析
2.1 不同发酵时期的理化性状分析
玉米秸秆好氧发酵不同时期堆料的理化性状如表1所示。建堆时(CS0)堆体平均温度34.0 ℃左右,第1次翻堆时(CS1)达到69.4 ℃,第2,3,4,5次翻堆后堆体温度仍均能够上升至55.0 ℃以上,说明好氧发酵时期堆体环境适宜,微生物活动旺盛。随着发酵的进行,堆料含水量呈下降趋势,从70.66%下降至58.00%。堆料pH值呈先下降后上升的趋势,由于前3次翻堆时堆体内微生物的活动导致有机酸的积累,pH值下降,后2次翻堆时堆体内氨化作用占优势,pH值又上升,在第5次翻堆时上升至8.13。E4/E6从建堆到第5次翻堆呈无规律变化,说明堆料在好氧发酵期间代表腐熟程度的胡敏酸含量尚不稳定。w(C)/w(N)在建堆和前3次翻堆时较高,后2次大幅降低,说明随着发酵的延长,堆体微生物可直接利用的水溶性碳源减少,水溶性氮相对增加。有机物含量则呈下降趋势,到第5次翻堆时,与建堆时相比下降了25.32个百分点。C/N值亦呈下降趋势,从建堆时的31.36下降至第5次翻堆时的22.86。以上结果表明,玉米秸秆条垛式好氧发酵正常,堆料的生物质转化过程比较顺利。
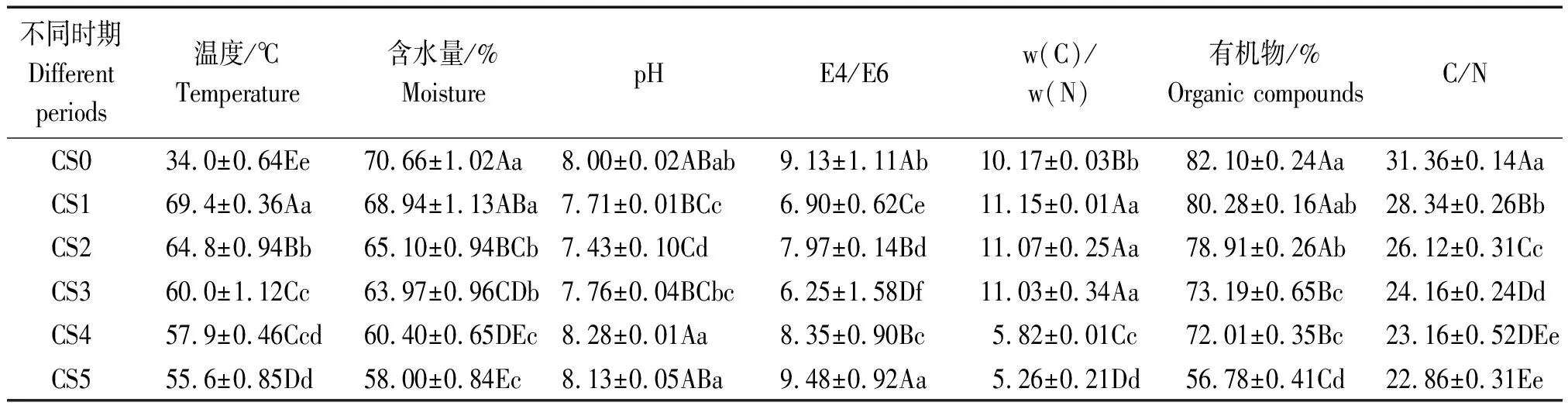
表1 不同好氧发酵时期堆料的理化性状Tab.1 Physicochemical characteristics of windrow during the different aerobic fermentation periods
注:不同大写字母表示处理间差异达极显著水平(P﹤0.01);不同小写字母表示处理间差异达显著水平(P﹤0.05)。
Note:Capital letters indicate very significant difference among different treatments;Lowercase letters indicate significant difference among different treatments.
2.2 细菌群落多样性分析
经PCR、高通量Illumina MiseqTM宏基因组16S rRNA测序、片段拼接和质控过滤后,获得秸秆发酵6个时期样品的有效序列为43 534~59 177条(表2)。由图1的细菌群落丰富度稀疏曲线可知,随着测序数量的增加,新的细菌菌系不断出现,Shannon指数均快速上升,当测序条带数量>20 000时,Shannon指数稀疏曲线趋于饱和,说明本研究的有效序列深度可用于描述整个细菌的群落结构。在97%的相似水平上对有效序列进行多样性分析,6个样品的文库覆盖率均达到99%,表明绝大部分的细菌种群都能被检测出,可以代表样本的真实情况。Chao1指数是用来估计群落中含OTU的总数目,由表2可知,用Chao1指数估计不同堆肥时期物种的OTU总数稍高。Shannon指数越大,群落多样性越高,所以不同时期的细菌多样性从高到低排序为CS5>CS3>CS1>CS2>CS0>CS4,CS5具有较高细菌群落多样性。Simpson指数越大,群落多样性越低,结合Simpson指数来看,CS5细菌群落多样性最高,CS1、CS2和CS3三者差异不明显。CS0和CS4均表现出较低的群落多样性,其中,以CS4群落多样性最低。
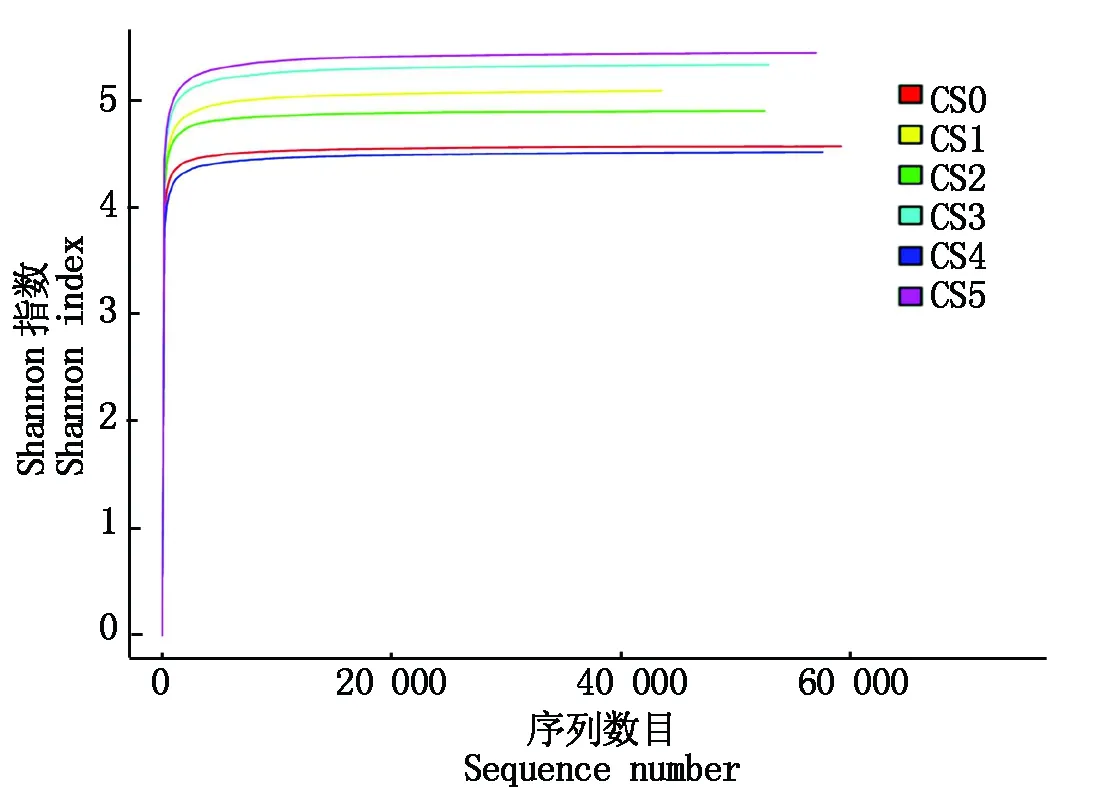
图1 细菌群落丰富度稀疏曲线Fig.1 Rarefaction Curve of the bacterial community abundance
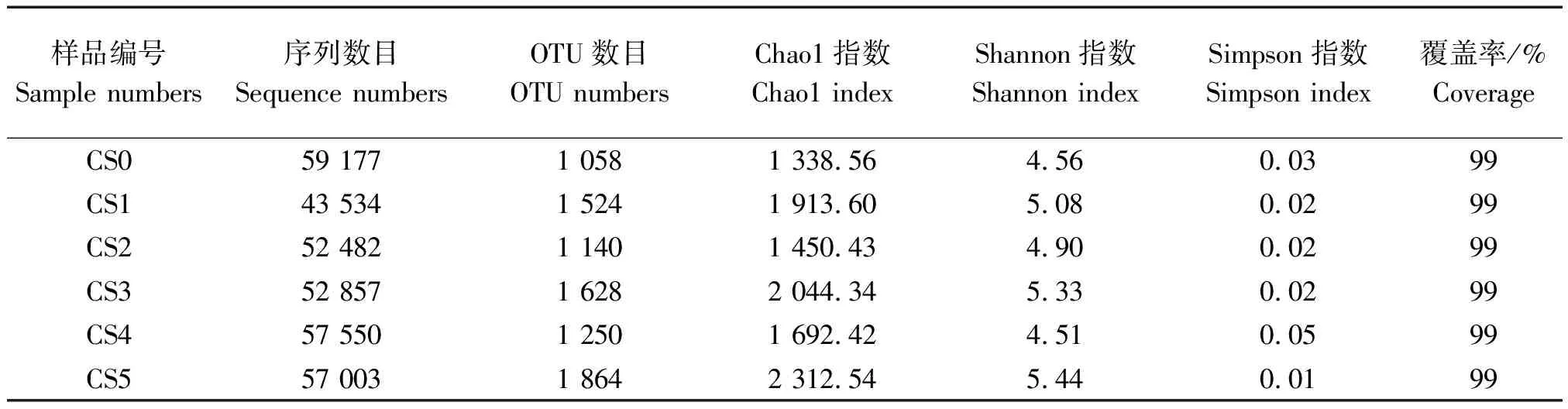
样品编号Sample numbers序列数目Sequence numbersOTU数目OTU numbersChao1指数Chao1 indexShannon指数Shannon indexSimpson指数Simpson index覆盖率/%CoverageCS059 1771 0581 338.564.560.0399CS143 5341 5241 913.605.080.0299CS252 4821 1401 450.434.900.0299CS352 8571 6282 044.345.330.0299CS457 5501 2501 692.424.510.0599CS557 0031 8642 312.545.440.0199
2.3 不同细菌群落结构在门水平的组成和相对丰度
在6个发酵时期,细菌随着发酵时间的延长菌群门类逐渐增多,在发酵初期(CS0),堆体内的微生物菌群丰度大于1%的优势门类仅有3种,到最后一次翻堆时(CS5)达到12种。好氧发酵期间共检测到细菌门类30种,优势门类主要为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)和放线菌门(Actinobacteria),它们的丰度之和在6个时期分别为88.88%,88.49%,91.88%,78.59%,58.62%和55.82%。4种门类在不同好氧发酵时期的菌群丰度不同,厚壁菌门为3.77%~75.69%,变形菌门为9.82%~35.72%,拟杆菌门为0.29%~36.00%,放线菌门为3.21%~13.83%。由表3可知,除了以上4种门类外,绿弯菌门(Chloroflexi)的丰度在CS4和CS5时期分别达到22.61%和16.30%。说明好氧发酵期间细菌菌群的种类和数量增多。

表3 细菌群落结构在门水平上的组成及相对丰度Tab.3 Bacteral community composition and relative abundance at phylum level %
2.4 不同堆肥时期细菌群落结构在目水平的组成和相对丰度
整个发酵期间共检测到78种细菌目类,6个不同时期共享的细菌目类有23种(将丰度均高于1%的菌目认为是共享菌目)。每个时期相对丰度排在前7位的优势目类不同(图2),CS0的优势目为芽孢杆菌目(Bacillales)22.84%、嗜热厌氧杆菌目(Thermoanaerobacterales)20.32%、放线菌目(Actinomycetales)13.28%、根瘤菌目(Rhizobiales)8.15%、黄色单胞菌目(Xanthomonadales)4.31%、梭菌目(Clostridiales)3.4%、黏球菌目(Myxococcales)1.61%;CS1的优势目为拟杆菌目(Bacteroidales)14.66%、芽孢杆菌目13.53%、假单胞菌目(Pseudomonadales)10.03%、梭菌目9.38%、根瘤菌目8.53%、黄色单胞菌目7.43%和鞘脂杆菌目(Sphingobacteriales)3.33%;CS2的优势目为梭菌目41.04%、芽孢杆菌目24.14%、放线菌目5.75%、嗜热厌氧杆菌目4.9%、黄色单胞菌目2.37%、厌氧绳菌目(Anaerolineales)1.93%和盐碱厌氧菌目(Natranaerobiales)1.76%;CS3的优势目为鞘脂杆菌目13.88%、黄色单胞菌目8.48%、拟杆菌目8.35%、噬纤维菌目(Cytophagales)8.31%、假单胞菌目6.23%、黄杆菌目(Flavobacteriales)5.23%和根瘤菌目3.52%;CS4的优势目为厌氧绳菌目21.43%、嗜热厌氧杆菌目9.09%、黄色单胞菌目9.09%、梭菌目8.23%、芽孢杆菌目5.23%、假单胞菌目4.78%和热脱硫杆菌目(Thermodesulfobacteriales)2.43%;CS5的优势目为厌氧绳菌目15.14%、鞘脂杆菌目7.38%、梭菌目5.98%、噬纤维菌目5.39%、热脱硫杆菌目4.36%、黄色单胞菌目4.05%和着色菌目(Chromatiales)3.16%。综上所述,6个阶段的共享优势目类(至少出现在3个时期)主要为芽孢杆菌目、梭菌目、黄色单胞菌目,嗜热厌氧杆菌目、根瘤菌目、假单胞菌目和厌氧绳菌目,但其丰度在各个时期不同,如芽孢杆菌目在发酵前期CS0、CS1和CS2中分别为22.84%,13.53%和24.14%,但在发酵后期CS3、CS4和CS5中仅有1.51%,5.23%和1.18%。
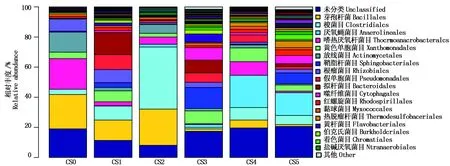
图2 不同发酵时期细菌群落结构在目水平上的组成和相对丰度Fig.2 Bacterial community composition and relative abundance at order level at different fermentation periods
2.5 不同堆肥时期细菌群落结构在属水平的组成和相对丰度

图3 不同发酵时期细菌群落结构在属水平上的组成和相对丰度Fig.3 Heatmap of bacteral community composition and relative abundance at genus level at different fermentation periods
本次发酵期,共检测到细菌属有644种,其中厚壁菌门83个属、变形菌门137个属、放线菌门47个属、拟杆菌门43个属,图3为部分菌属的热图。不同发酵时期共享的主要属(同种属至少在3个样品中相对丰度不小于1%)为Povalibacter、梭状芽孢杆菌属(ClostridiumⅢ)、芽孢杆菌属(Bacillus)、赖氨酸芽孢杆菌属(Lysinibacillus)、嗜热脲芽孢杆菌属(Ureibacillus)、假单胞菌属(Pseudomonas)和Chryseolinea。它们的高丰度体现在不同时期,Povalibacter在整个发酵时期丰度均较高,芽孢杆菌属、赖氨酸芽孢杆菌属和嗜热脲芽孢杆菌在建堆初期CS0和前2次翻堆(CS1和CS2)时较高,到后3次翻堆时下降至1%以下。梭状芽孢杆菌属丰度在建堆初期低于1%,但在第2次翻堆时达到15.57%。假单胞菌属和Chryseolinea在发酵前期丰度较低,后期丰度较高。结果显示,好氧发酵不同时期影响不同种类细菌的菌群丰度。
在属水平上对不同时期进行聚类分析,结果显示,CS3和CS5距离最近,CS0和CS4距离最近,CS1分别与CS3和CS5二者以及CS0和CS4二者之间距离较近,CS2与它们的距离最远,说明CS3和CS5细菌菌群相似,CS0和CS4的细菌菌群相似,CS2与其他时期的细菌菌群距离最远、相似性最差(图3)。
3 讨论与结论
3.1 好氧发酵时期的理化性状
好氧发酵过程中实时监测堆料的理化性状对于堆肥品质的掌控非常关键。堆料的初始w(C)/w(N)、含水量和pH值决定着堆体发酵能否正常启动,这3个因素为微生物提供了必需的营养和生长条件。本研究中,堆体温度快速上升,在发酵期间堆体深度45 cm处温度均能达到55.0 ℃以上,说明好氧发酵正常,微生物活动旺盛,适时翻堆通气是保持较高温度的原因。微生物活动和繁殖所要求的适宜C/N值一般为25∶1,由于受到堆料原始组成结构的影响,全部碳素并没有被微生物直接快速利用,所以C/N稍高,可溶性的碳素就越多,从而有利于堆体发酵的启动,合适的C/N对好氧发酵的启动至关重要。一般认为,堆肥材料中初始C/N在26∶1~35∶1较为适宜[16]。E4/E6值能反映胡敏酸分子的稳定程度和腐殖化程度,E4/E6值越小,表示分子中芳香环的缩合度、芳构化度和分子量均较大,且平均存留时间越长[17-18]。本研究中,到第5次翻堆时E4/E6仍较高,说明此时堆体仍处于矿化阶段,腐殖化程度还较低。
3.2 影响发酵期间的细菌群落结构的因素
本研究在好氧发酵期间共检测到30种细菌门类、78种细菌目类和644种已命名的细菌属类,厚壁菌门、拟杆菌门、变形菌门和放线菌门4个主导菌门的总丰度在6个发酵时期均较高。Antunes等[19]研究表明,堆肥高温期的细菌分类优势门类占总分类reads的85%。张丽丽[20]同样用玉米秸秆自然堆肥,共鉴定到了24个细菌门类,4个主导门类共占到样品总分类reads的93.5%。表明厚壁菌门、拟杆菌门、变形菌门和放线菌门在堆肥好氧发酵阶段作用最强,同时说明堆料配比及堆肥工艺会影响堆体内的细菌门类及其相应丰度。
细菌群落的结构组成会受到堆体高温和通气性的共同影响。张丽丽[20]利用发酵罐控制堆肥温度和氧浓度,可以明显提高堆肥中放线菌门和厚壁菌门的含量,并通过玉米秸秆堆肥试验,发现在20~50 cm深度放线菌门会快速生长。贾洋洋[10]在秸秆堆肥生境的研究也表明,微生物白色菌丝生长旺盛的区位有着更高的胞外纤维素酶活性,具有更高的纤维素降解率,说明放线菌偏爱氧浓度较高的堆体深度。放线菌门之前被认为是堆肥嗜热阶段占据主导地位的细菌门类[21],从本研究看,放线菌门类丰度虽然为主导门类,但丰度并不太高,可能与堆体供氧不足有关,所以及时翻堆是保证堆体供氧充足的有效措施[22]。有研究表明,堆肥过程中厚壁菌门的高丰度主要是其耐热性和兼性厌氧性[23-24],绿弯菌门也是一种兼性厌氧生物[25]。本研究中,厚壁菌门和绿弯菌门在发酵期间出现较高的比例,推测是由于本次发酵堆料含水量偏高,堆体偏紧实,内部缺氧,兼性厌氧菌发挥作用的结果。高温对拟杆菌门和变形菌门的生长有抑制作用,相对较低的温度下更适合这2种门类的生长[26],本研究堆体内部缺氧,温度较低,可能是变形菌门和拟杆菌门所占比例较大的原因。
3.3 好氧发酵期间的细菌群落结构及菌群演替
不同发酵时期堆料微生物群落组成不同。张丽丽[20]研究结果表明,在堆肥初期时,占优势的主要菌属为拟杆菌门和变形菌门,包括Petrimonas、Proteiniphilum和假单胞菌属等,但到堆肥后期这些菌属被厚壁菌门的芽孢杆菌属和放线菌门的喜热裂孢菌属等所取代。本研究中,变形菌门的Povalibacter在整个发酵期均有较高的丰度,厚壁菌门的芽孢杆菌属、赖氨酸芽孢杆菌属、嗜热脲芽孢杆菌属以及高温单孢菌属均在发酵前期丰度较高,发酵后期丰度较低。而厚壁菌门的梭状芽孢杆菌属、变形菌门的假单胞菌属和拟杆菌门的Chryseolinea在发酵后期出现较高丰度,说明细菌群落随着发酵进程发生了明显的群落演替现象,起作用的菌属类别及其相对丰度也发生了变化。
3.4 好氧发酵期间与木质纤维素降解有关的菌群
厚壁菌门、变形菌门、拟杆菌门和放线菌门被认为是在木质纤维素堆肥中主导的细菌门类,其中,放线菌门和厚壁菌门是好氧堆肥中主要的木质纤维素降解微生物群落[14,20],这2种门类在本研究中丰度较高,说明它们在玉米秸秆降解中起到关键作用。已有报道,厚壁菌门的热杆菌属(Thermobacillus)、放线菌门喜热裂孢菌属(Thermobifida)、嗜热多孢菌属(Thermopolyspora)、高温单孢菌属(Thermomonospora)、变形菌门假黄单胞菌属(Pseudoxanthomonas)和纤维弧菌属(Cellvibrio)是与木质纤维素降解有关的菌属[10,20,27]。Guo等[28]研究表明,在以麦草为主料的堆肥过程主要的优势种属包括芽孢杆菌属、类芽孢杆菌属(Paenibacillus)、高温单孢菌属、嗜热梨囊鞭菌属(Thermasporomyces)、假单胞菌属和纤维弧菌属(Cellvibrio)等。芽孢杆菌属具有耐热性,是木质纤维素堆肥中厚壁菌门的一个常被鉴定到的属[29-31]。但是在本研究中,热杆菌属、喜热裂孢菌属、嗜热多孢菌属、高温单孢菌属和类芽孢杆菌属均是在建堆初期丰度较高,分别为2.29%,1.46%,2.41%,3.67%和1.15%,其他发酵时期丰度均较低。纤维弧菌属建堆初期未检测到,但在第3次翻堆时丰度达到4.52%。综合以上说明好氧发酵不同时期和不同配比的堆料,出现木质纤维素降解菌属的种类和丰度不同[29-33]。本研究以玉米秸秆为主料的好氧发酵过程中,在6个发酵时期检测到的共享属有Povalibacter、梭状芽孢杆菌属、芽孢杆菌属、赖氨酸芽孢杆菌属、嗜热脲芽孢杆菌属、假单胞菌属和Chryseolinea,其中,变形菌门黄色单胞菌目的Povalibacter、厚壁菌门芽孢杆菌目的梭状芽孢杆菌属和赖氨酸芽孢杆菌属以及拟杆菌门噬纤维素菌目的Chryseolinea尚未见与木质纤维素降解有关的报道。所以,从这些菌属中有可能筛选出与加快玉米秸秆快速腐熟的新菌种,下一步结合以PCR为基础的DGGE等分子生物学技术进行各种降解功能菌株的分离研究,通过菌剂添加用于好氧发酵堆肥,将有助于加快堆肥腐熟和改善堆肥品质。
堆料初始C/N、含水量以及pH值决定着玉米秸秆好氧发酵能否正常启动,而及时翻堆可以确保堆料混合均匀,保证氧气供应充分,微生物活动正常,使堆体正常升温至55.0 ℃以上。本研究结果表明,玉米秸秆好氧发酵不同时期所涉及的细菌菌门、菌目和菌属的数目及其丰度不同,表现出明显的细菌群落结构多样性和菌群演替现象。玉米秸秆不同发酵期间共享的优势门类为厚壁菌门、变形菌门、拟杆菌门和放线菌门,共享的主要优势目类为芽孢杆菌目、梭菌目、黄色单胞菌目、嗜热厌氧杆菌目、根瘤菌目、假单胞菌目和厌氧绳菌目,共享的主要属为Povalibacter、梭状芽孢杆菌属、芽孢杆菌属、赖氨酸芽孢杆菌属、嗜热脲芽孢杆菌属、假单胞菌属和Chryseolinea,它们是玉米秸秆好氧发酵的关键性细菌类别,为进一步筛选与玉米秸秆快速腐熟的新菌种指明了研究方向,并提供了参考依据。
参考文献:
[1] 张永锋,滕 星,李忠和,等. 玉米秸秆堆肥及其影响因素研究进展[J]. 吉林农业大学学报,2016,38(5):613-618.
[2] 石建森. 玉米秸秆基质压缩块栽培双孢菇[J]. 农村百事通,2013,40(19):42-43.
[3] 郭金岭,智利红,张 歌. 玉米秸秆基质对无土栽培莴苣生长的影响[J]. 北方园艺,2011(13):34-35.
[4] 刘 洋,李爱华. 玉米秸秆堆制有机肥对植烟土壤理化性状的影响[J]. 作物研究,2016,30(2):149-151.
[5] Amann I,Ludwig W,Schleifer H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J]. Microbiological Review,1995,59(1):143-169.
[6] 马海霞,张丽丽,孙晓萌,等. 基于宏组学方法认识微生物群落及其功能[J]. 微生物学通报,2015,42(5):902-912.
[7] 李 慧,何晶晶,张 颖,等. 宏基因组技术在开发未培养环境微生物基因资源中的应用[J]. 生态学报,2008,28(4):1762-1773.
[8] 李 洁,李睿玉,杨 红,等. 土壤微生物多样性的研究方法[J]. 山西农业科学,2016,44(11):1738-1742,1746.
[9] 冯明谦,刘德明. 滚筒式高温堆肥中微生物种类数量的研究[J]. 中国环境科学,1999,19(6):490-492.
[10] 贾洋洋. 利用宏基因组方法分析堆肥生境中微生物区系的变化[D]. 济南:山东大学,2012.
[12] De Gannes V,Eudoxie G,Hickey W J. Prokaryotic successions and diversity in composts as revealed by 454-pyrosequencing[J]. Bioresource Technology,2013,133:573-580.
[13] 沈其荣,王瑞宝,王 岩. 堆肥制作中的生物化学变化特征[J]. 南京农业大学学报,1997,20(2):51-57.
[14] Partanen P,Hultman J,Paulin L,et al. Bacterial diversity at different stages of the composting process[J]. BMC Microbiology,2010,10:94.
[15] 刘雯雯. 利用菌糠制作生物有机菌肥的途径及其效果研究[D]. 兰州:甘肃农业大学,2008.
[16] 范长征. 堆肥过程中木质素降解及甲烷排放相关功能基因研究[D]. 长沙:湖南大学,2015.
[17] 文启孝. 土壤有机质研究法[M]. 北京:农业出版社,1984.
[18] 鲍艳宇,周启星,娄翼来,等. 奶牛粪好氧堆肥过程中不同含碳有机物的变化特征以及腐熟评价[J]. 生态学杂志,2010,29(11):2111-2116.
[19] Antunes L P,Martins L F,Pereira R V,et al. Microbial community structure and dynamics in thermophilic composting viewed through metagenomics and metatranscriptomics[J]. Scientific Reports,2016,6:38915.
[20] 张丽丽. 整合宏组学方法揭示天然木质纤维素堆肥中的关键功能微生物群落[D]. 济南:山东大学,2016.
[21] Piceno Y M,Pecora-Black G,Kramer S,et al. Bacterial community structure transformed after thermophilically composting human waste in Haiti[J]. PLoS One,2017,12(6):e0177626.
[22] Villar I,Alves D,Mato S. Seafood-Processing sludge composting:changes to microbial communities and Physico-Chemical parameters of static treatment versus for turning during the maturation stage[J]. PLoS One,2016,11(12):e0168590.
[23] Zhang X,Zhong Y,Yang S,et al. Diversity and dynamics of the microbial community on decomposing wheat straw during mushroom compost production[J]. Bioresource Technology,2014,170:183-195.
[24] Tian W,Sun Q,Xu D B,et al. Succession of bacterial communities during composting process as detected by 16S rRNA clone libraries analysis[J]. International Biodeterioration & Biodegradation,2013,78:58-66.
[25] Wang C,Dong D,Wang H,et al. Metagenomic analysis of microbial consortia enriched from compost:new insights into the role of actinobacteria in lignocellulose decomposition[J]. Biotechnology for Biofuels,2016,9:22.
[26] Kadnikov V V,Mardanov A V,Podosokorskaya O A,et al. Genomic analysis ofMelioribacterroseus,facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi group[J]. PLoS One,2013,8(1):e53047.
[27] Székely A J,Sipos R,Berta B,et al. DGGE and T-RFLP analysis of bacterial succession during mushroom compost production and sequence-aided T-RFLP profile of mature compost[J]. Microbial Ecology,2009,57(3):522-533.
[28] Guo Yaping,Zhang Guoqing,Chen Qingjun,et al.Analysis on bacterial community structure in Mushroom(Agaricusbisporus)compost using PCR-DGGE[J]. Agricultural Science & Technology,2015,20(8):1778-1784.
[29] 梁军锋,张洪生,张克强,等. 木质素降解菌的筛选及对秸秆的降解研究[J]. 华北农学报,2009,24(5):206-209.
[30] Mayende L,Wilhelmi B S,Pletschke B I. Cellulases(CMCases)and polyphenol oxidases from thermophilicBacillusspp. isolated from compost[J]. Soil Biology & Biochemistry,2006,38(9):2963-2966.
[31] 王士强,顾春梅,赵海红. 木质纤维素生物降解机理及其降解菌筛选方法研究进展[J]. 华北农学报,2010,5(S1):313-317.
[32] 陈婷婷,王丽芳,王 琪,等. 芦笋老茎堆肥中嗜热放线菌的分离和鉴定[J]. 山西农业科学,2013,41(1):70-74.
[33] Chandna P,Nain L,Singh S,et al. Assessment of bacterial diversity during composting of agricultural byproducts[J]. BMC Microbiology,2013,13:99.
