银屑病发病机制相关信号通路研究进展
2018-07-02姜蔚蔚张春雷
姜蔚蔚 张春雷
银屑病是一种临床表现为红斑、斑块、鳞屑的慢性炎症性皮肤病;表皮角化过度、角化不全、棘层增厚和真皮炎症是其组织病理特征。其发病机制至今不清,目前认为与遗传、环境和免疫等多因素相关。全基因组关联研究显示,超过20个易感位点均参与银屑病的发病。目前认为银屑病是一个累及多环节多通路的疾病,以下就现有关于银屑病发病机制中的信号通路及其相关调节器的研究做一综述,为其进一步靶向治疗的研究探索思路。
1 炎症介导途径
1.1 IL-23/IL-17A-Th17轴(Interleukin-23/Interleukin-17A-T Helper type 17 cells axis) 基于临床、实验和基因方面的研究发现由IL-23发挥主要诱导功能的Th17细胞介导的慢性炎症在银屑病的发病机制中起重要作用[1]。IL-23在调节Th17细胞终端效应分子的功能上扮演重要角色,主要包括维持Th17细胞的功能及其促炎因子的产生,而发挥此功能的主要原因则是保护性的IL23R381等位基因的存在[2],IL-23与受体的结合产生的相关信号同时也刺激了下游STAT-3通路的活化[3]。近年来又有研究表明,IL-17A在银屑病样炎症小鼠模型中也起诱导表皮增生的作用[4],在今后的研究中可将IL23R作为预测治疗效果的表面信号分子。
早期生长反应因子-1(Early growth response-1,Egr-1)是一种在细胞生长、分化、生存和免疫反应中起重要作用的转录因子。已发现Egr-1在银屑病皮损中高表达,现有研究通过微阵列分析发现,在银屑病患者和体外培养的人角质形成细胞中均显示IL-17A导致Egr-1的高表达,且IL-17A诱导的Egr-1的表达可被细胞外信号调节激酶(Extracellular signal-regulated kinase,ERK)抑制剂抑制,针对Egr-1的小分子干扰RNA可导致IL-17A诱导的银屑病素下调,证明Egr-1可能是IL-17A介导银屑病免疫反应的潜在的重要调节器[5]。
最近有研究发现,转录辅激活物IkappaBzeta在IL-17A介导的免疫效应中起重要作用,提示IkappaBzeta可能是银屑病发病中的一个新的调节机制,在未来银屑病或其他IL-17A介导的疾病靶向治疗中有广阔的研究前景[6]。
1.2 IFN-γ/TNF-Th1轴(interferon-γ/Tumor necrosis factor-T Helper type 1 cells axis) IFN-γ以及随后出现的IL-17A/Th17轴被认为是参与银屑病机制的中心细胞因子。早期一些研究数据表明,IFN-γ诱导了STAT-1和p48两条信号通路[7]。后来又有研究证实,有生物活性的sTNF(可溶TNF)和tmTNF(跨膜TNF)与相应受体结合启动了三个主要的免疫调控通路,首先是NF-kappaB活化和炎症的产生,其次是MAPK(促分裂原活化蛋白激酶)通路和癌基因氨基端激酶的活化,随后促进细胞的分化、增殖和凋亡,最后则与死亡信号有关[8,9]。细胞的生长、幸存和凋亡是在同一个细胞内通过同一个细胞因子的凋亡和抗凋亡信号共同刺激活化而实现的,在银屑病的发病中起重要作用。
1.3 TGF-β(Transforming growth factor,TGF-β1)信号通路 TGF-β信号可通过其下游的Smad依赖和非依赖通路发挥生物效应。据报道,在动物模型中其过表达导致了银屑病样皮损,关于发生此生物作用的具体机制,有研究发现,在TGF-β转基因的小鼠中,给予Smad抑制剂刺激,则会导致银屑病皮损明显改善以及相关浸润细胞因子的减少,所以认为TGF-β介导银屑病样皮损是通过Smad依赖途径,且应用Smad抑制剂靶向阻断TGF-β/Smad信号通路可能是银屑病的一个新的治疗靶点[10]。
去泛素化酶泛素特定肽酶15(ubiquitin specific peptidase-15, USP15)是TGF-β信号通路的调节剂,整个通路需要依赖TGF-β信号的抑制因子Smad7以及TGF-β受体1(TbetaR-I)实现。USP15在许多肿瘤的机制中被认为发挥重要作用,有研究发现在银屑病皮损和细胞系中USP15和Smad7的表达高度正相关,而USP15与TbetaR-I以及Smad7与TbetaR-I的表达呈负相关,同时在Hacat细胞系中转染USP15则导致TbetaR-mRNA上调以及Smad7的下调,可以认为USP15在银屑病的发病中起重要作用,且可能通过调控TbetaR-I/Smad7或其他通路实现[11]。
2 激酶传导途径
2.1 JAK/STAT(Janus kinase/signal transducer and activator of transcription)酪氨酸激酶信号转导及转录激活蛋白 大量研究显示此信号通路参与包括造血和免疫反应在内的多种生理过程,调节JAK/STAT信号的基因突变可导致多种炎症反应的失调以及骨髓增生性疾病,同时此信号通路及相关细胞因子在银屑病发病机制中也被证实有至关重要的作用,可能是一个潜在的新的治疗靶点[12]。JAK/STAT信号通路在银屑病中发挥作用的主要为JAK/STAT1和JAK/STAT3,调控通路的细胞因子包括IFN-γ、IL-17、IL-22、IL-6等,且此通路不仅在T细胞中发挥作用,在角质形成细胞中也发挥重要作用。JAK/STAT3通路抑制剂枸橼酸托法替布在银屑病的应用已完成相关临床试验。银屑性是一种T细胞介导的炎症反应,JAK3主要表达在T细胞中,研究证实[13]在银屑病样小鼠中选择性阻断JAK3信号可导致临床上银屑病皮损减轻及银屑病组织标志物的减少,引起导致银屑病相关细胞因子如IL-17、IL-22、IL-23以及TNF-α等细胞因子的显著降低[14-16]。
IL-22是由不同种类的淋巴细胞(如活化T细胞,Th17、Th22、NK细胞等)产生的IL-10细胞因子家族的成员,其发挥功能主要是通过IL22-IL22R复合物的形成以及随后的JAK传感器和JAK-STAT信号通路的活化。有研究证实其在包括银屑病在内的多种自身免疫性疾病中有至关重要的作用[17],其主要通过抗菌蛋白促进形成生物屏障以及干扰终端角质层细胞的分化、募集中性粒细胞诱导细胞因子的产生等方式调节角质形成细胞的功能[18]。
有研究将豚鼠分为银屑病组和正常组,用流式细胞分析技术分别检测外周血中的Th17/IL-17的比例,同时运用免疫组化和蛋白印迹检测STAT和血管内皮生长因子(Vascular endothelial growth factor,VEGF)的浓度,结果银屑病组Th17的表达显著高于正常对照组,Th17相关因子STAT3和VEGF也较正常对照组显著升高,和Th17的表达正相关,结果提示Th17细胞在银屑病的发病中发挥作用可能是通过STAT3-VEGF通路实现的[19]。
微囊蛋白-1(caveolin-1,CAV-1)参与信号转导的调节,异常的CAV-1表达参与很多疾病的发病,关于是否参与银屑病的发病,有研究发现银屑病患者皮损表皮中CAV-1的表达显著降低;而使体外角质形成细胞中CAV-1基因沉默则显示出STAT-3信号通路强烈的活化,以及角蛋白16和几种银屑病相关细胞因子(如TNF-α)的高表达;此外,在银屑病样小鼠模型中控制CAV-1区域肽段则会改善皮肤表型、降低表皮厚度和浸润细胞计数,且相关细胞因子TNF-α、IL17A和IL23等均显著被抑制。综上研究证实CAV-1可能通过STAT3信号通路和细胞因子网络在银屑病的发病机制中起作用[20]。
2.2 MAPK(Mitogen-activated protein kinase)丝裂原激活的蛋白激酶通路 趋化因子样因子-1(Chemokine-like factor 1,CKLF1)被认为参与银屑病的局部炎症和细胞增殖。有研究发现银屑病患者皮损中CKLF1及其受体CCR4的表达均升高,且在其诱导的C端肽(C19和C27)的刺激下,人原始脐静脉内皮细胞显示高度增殖,此外,肽的增强作用还伴随着ERK1/2-MAPK信号通路的活化,因此我们认为CKLF1通过促进微血管内皮细胞的增殖参与银屑病的发生,且可能是通过ERK1/2-MAPK的活化来实现的[21]。
另外,新发现的数据显示p38 MAPK、ERK1/2(Extracellular signal-regulated kinase1/2)和JNK(c-Jun N-terminal kinase)在银屑病的发生发展中发挥潜在作用[22]。
2.3 PI3K/mTOR(Phosphatidyl Inositol 3-kinase/Mammalian Target Of Rapamycin)/Akt磷脂酰肌醇-3-羟激酶激酶/雷帕霉素靶蛋白/蛋白激酶B信号通路 肿瘤抑制磷酸酶和张力蛋白同族体(Tumor suppressor phosphatase and tensin homolog,PTEN)可通过PI3K/Akt信号通路抑制细胞的增殖,其功能缺失和突变在许多肿瘤中得到验证。在银屑病的研究中发现,相较于正常皮肤对照组,银屑病患者皮损组PTEN的mRNA水平降低,PTEN蛋白也低表达,结合前期有研究证实,银屑病皮损中Akt的活化高于正常对照组,得出结论,PTEN的下调可能通过在银屑病皮损中过度活化PI3K/Akt信号通路发挥作用,与银屑病角质形成细胞的过度增殖密切相关[23]。
现有证据表明,中枢神经系统与免疫系统在银屑病的发病机制中存在双向作用,而皮肤神经系统在其中的作用却尚未明了。近期有研究发现,将神经生长因子(Nerve growth factor, NGF)作用于银屑病样细胞模型中,出现低氧诱导因子-1α(Hypoxia-inducible factor-qα)和VEGF的升高,而且mTOR抑制物雷帕霉素预处理之后不会出现上述数据的走高,研究认为,NGF在银屑病样细胞系中调控VEGF的表达是通过PI3K/mTOR信号通路实现的[24]。此外,关于PI3K/Akt/mTOR信号通路在银屑病的发病机制中出现越来越多的研究,许多针对细胞间信号的小分子进行预处理表现出显著的有效性。众多数据表明PI3K/Akt/mTOR信号通路可能是银屑病的一个潜在治疗靶点[25]。
3 核内基因转录水平途径
3.1 NF-KappaB(Nuclear transcription factor kappa B)核转录因子通路 核转录因子是一个可以精细调节多种炎症和复杂生物过程的蛋白转录因子,也是多种免疫和炎症通路的关键调节因素,在细胞的增殖、分化和凋亡中都起重要作用,而此重要作用主要是通过调节编码促炎调节因子、扩大和维持慢性炎症反应以及抗凋亡等多种类的基因实现的,其始动于一些促炎刺激物,如TNF、IL-1等的激发。银屑病也是一种慢性炎症性皮肤病,并以高水平活化的磷酸化的NF-kappaB为主要标志,基因组专家也经研究证实银屑病和NF-kappaB通路的调控息息相关,并与其角质形成细胞和免疫细胞行为的改变联系起来,随后也出现了针对此通路的银屑病靶向治疗方法[26]。
水通道蛋白(Aquaporin3, AQP3)是一种水/甘油通道蛋白,被发现可以转运H2O2,有研究发现敲除AQP3的银屑病样小鼠可使IL-23诱导的银屑病样皮损减轻,同时伴随NF-kappaB通路活化以及细胞间H2O2积聚的异常,而H2O2起调节NF-kappaB活化的作用,这些数据表明,银屑病中TNF-α诱导的NF-kappaB通路依赖于AQP3的表达[27]。
胱冬肽酶募集结构域家族成员CARD14基因的突变被认为与银屑病的发病相关。CARD14是一种调节NF-kappaB活化的折叠蛋白,而与银屑病相关的CARD14的突变导致NF-kappaB信号的增强。有研究运用免疫荧光技术证明,CARD14高表达于CD31+的内皮细胞,同时在CARD14+CD31+的内皮细胞中也发现了磷酸化的NF-kappaB,所以认为CARD14在内皮细胞中通过NF-kappaB信号通路的表达对银屑病中增加细胞因子的表达和免疫细胞的招募起重要作用[28]。
富半胱氨酸61(Cysteine-rich 61,Cyr61/CCN1)是一种细胞外基质蛋白,也是一种高度表达在银屑病皮损中的促炎因子,有研究表明,阻断CCN1的功能可以引起银屑病样小鼠模型表皮增生和炎症减轻,同时CNN1可导致正常人角质形成细胞和细胞系的活化,包括细胞增殖和免疫相关分子的表达,并且CCN1的刺激还活化了下游的磷酸肌醇-3激酶/Akt/NF-kappaB信号通路[29]。
β-转导重复相容蛋白(Beta-transducin repeat-containing protein,betaTrCP)是一种识别泛素连接酶组成成分的基质,可以控制重要信号通路调节器的稳定性,如银屑病的关键炎症信号通路-NF-kappaB信号通路。有研究显示,咪喹莫特诱导的银屑病样小鼠模型中betaTrCP水平升高,kappaB抑制物降低,NF-kappaB活化增强,而且在TNF-α诱导的银屑病样细胞模型和正常人角质形成细胞中敲除betaTrCP可以显著抑制NF-kappaB信号通路的过度活化和细胞间粘附分子1(ICAM-1)的表达,可以认为,betaTrCP可能参与NF-kappaB信号通路介导的银屑病相关炎症反应,同时也是一个潜在的治疗靶点[30]。
3.2 Wnt信号通路 分泌型卷曲相关蛋白4(secreted frizzled-related protein-4,SFRP-4)是Wnt信号通路的负性调控因子,在银屑病动物模型及患者皮损中均表达降低,SFRP4直接抑制过量角质形成细胞的增殖,给予Wnt信号通路的抑制剂或体内注射SFRP4可改善银屑病的皮肤表现,包括炎症细胞的浸润等组织病理改变,可以认为SFRP的下调可能是银屑病的发病机制之一[31]。
3.3 TLR7(Toll-like receptor7)介导的信号通路 腺病毒N端24氨基酸绿色荧光蛋白(adenovirus Nterminal 24 amino acids green fluorescent protein, ADN24GFP)有效抑制内毒素诱导的相关细胞因子的表达,p55磷脂酰肌醇3激酶(p55 regulatory subunit, p55PIK)的高表达与内毒素协同作用促进炎症因子的释放,且在体外银屑病样细胞中发现,ADN24GFP和p55PIKGFP影响内毒素诱导炎症因子释放的作用是通过TLRs/MyD88信号通路实现的[32]。
此外,TLR1,2在银屑病患者皮损中高表达,TLR4在点滴型银屑病中表达高于斑块型和正常对照人群,TLR5,9在银屑病角质形成细胞中通过TGF-α而上调,另外Begon等发现银屑病角质形成细胞中TLR2,3,4信号通路诱导的TNF-α和IL-8的表达是通过NF-kB核转位实现的。基于以上研究事实,TLR基因在银屑病的发病机制中可能扮演重要角色[33]。
总之,银屑病作为一种以遗传、免疫和环境为基础多因素参与的复杂疾病,其信号通路错综复杂,环环相扣(图1)。本文希望通过对几大主要通路的简要梳理,对银屑病的发病机制有个更为宏观的认识,为之后更深入的机制探索理清思路。
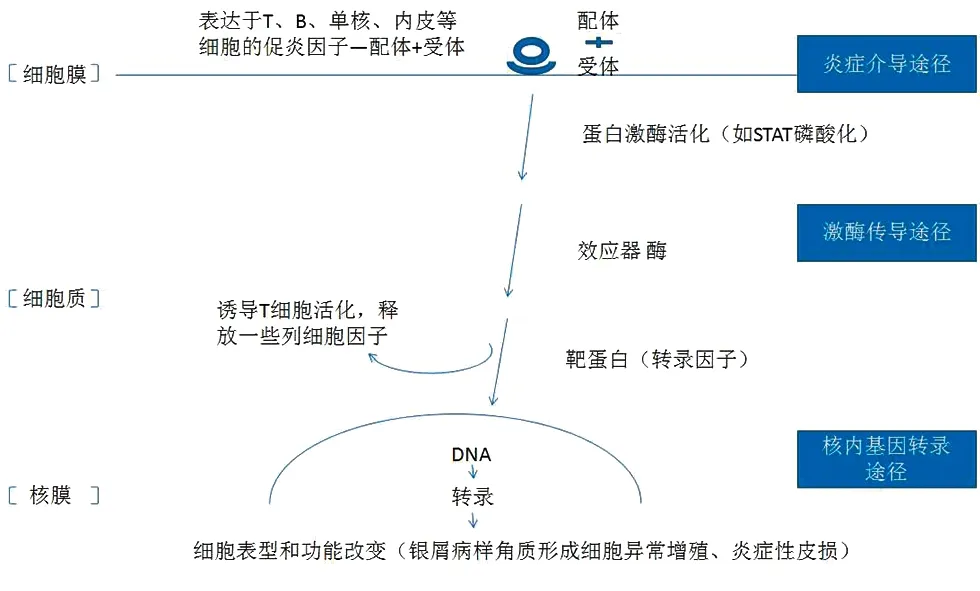
图1 银屑病信号通路宏观梳理概括
[1] Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis[J]. J Invest Dermatol,2009,129(6):1339-1350.
[2] Perera GK, Di Meglio P, Nestle FO. Psoriasis[J]. Annu Rev Pathol,2012,7:385-422.
[3] Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation[J]. Annu Rev Immunol,2007,25:221-242.
[4] Rizzo HL, Kagami S, Phillips KG, et al. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A[J]. J Immunol,2011,186(3):1495-1502.
[5] Jeong SH, Kim HJ, Jang Y, et al. Egr-1 is a key regulator of IL-17A-induced psoriasin upregulation in psoriasis[J]. Exp Dermatol,2014,23(12):890-895.
[6] Johansen C. IκBζ: A key protein in the pathogenesis of psoriasis[J]. Cytokine,2016,78:20-21.
[7] Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN[J]. FASEB J,1999,13(3):495-502.
[8] Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily[J]. Biochem Pharmacol,2003,66(8):1403-1408.
[9] Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology[J]. Cell,2001,104(4):487-501.
[10] Zhang Y, Meng XM, Huang XR, et al. Transforming growth factor-β1 mediates psoriasis-like lesions via a Smad3-dependent mechanism in mice[J]. Clin Exp Pharmacol Physiol,2014,41(11):921-932.
[11] Feng AP, He YM, Liu XX, et al. Expression of USP15, TβR-I and Smad7 in psoriasis[J]. J Huazhong Univ Sci Technolog Med Sci,2014,34(3):415-419.
[12] Palanivel JA, Macbeth AE, Chetty NC, et al. An insight into JAK-STAT signalling in dermatology[J]. Clin Exp Dermatol,2014,39(4):513-518.
[13] Chang BY, Zhao F, He X, et al. JAK3 inhibition significantly attenuates psoriasiform skin inflammation in CD18 mutant PL/J mice[J]. J Immunol,2009,183(3):2183-2192.
[14] Boy MG, Wang C, Wilkinson BE, et al. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patients with psoriasis[J]. J Invest Dermatol,2009,129(9):2299-2302.
[15] Papp KA, Menter A, Strober B, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study[J]. Br J Dermatol,2012,167(3):668-677.
[16] Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis[J]. Br J Dermatol,2013,169(1):137-145.
[17] Yang X, Zheng SG. Interleukin-22: a likely target for treatment of autoimmune diseases[J]. Autoimmun Rev,2014,13(6):615-620.
[18] Sabat R, Wolk K. Research in practice: IL-22 and IL-20: significance for epithelial homeostasis and psoriasis pathogenesis[J]. J Dtsch Dermatol Ges,2011,9(7):518-523.
[19] Zheng XF, Sun YD, Liu XY. Correlation of expression of STAT3, VEGF and differentiation of Th17 cells in psoriasis vulgaris of guinea pig[J]. Asian Pac J Trop Med,2014,7(4):313-316.
[20] Yamaguchi Y, Watanabe Y, Watanabe T, et al. Decreased Expression of Caveolin-1 Contributes to the Pathogenesis of Psoriasiform Dermatitis in Mice[J]. J Invest Dermatol,2015,135(11):2764-2774.
[21] Tan Y, Wang Y, Li L, et al. Chemokine-like factor 1-derived C-terminal peptides induce the proliferation of dermal microvascular endothelial cells in psoriasis[J]. PLoS One,2015,10(4):e0125073.
[22] Mavropoulos A, Rigopoulou EI, Liaskos C, et al. The role of p38 MAPK in the aetiopathogenesis of psoriasis and psoriatic arthritis[J]. Clin Dev Immunol,2013,2013:569751.
[23] Li Y, Man X, You L, et al. Downregulation of PTEN expression in psoriatic lesions[J]. Int J Dermatol,2014,53(7):855-860.
[24] Zhang J, Ma WY. Nerve growth factor regulates the expression of vascular endothelial growth factor in human HaCaT keratinocytes via PI3K/mTOR pathway[J]. Genet Mol Res,2014,13(4):9324-9335.
[25] Huang T, Lin X, Meng X, et al. Phosphoinositide-3 kinase/protein kinase-B/mammalian target of rapamycin pathway in psoriasis pathogenesis. A potential therapeutic target?[J]. Acta Derm Venereol,2014,94(4):371-379.
[26] Goldminz AM, Au SC, Kim N, et al. NF-κB: an essential transcription factor in psoriasis[J]. J Dermatol Sci,2013,69(2):89-94.
[27] Hara-Chikuma M, Satooka H, Watanabe S, et al. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-κB signalling in keratinocytes and development of psoriasis[J]. Nat Commun,2015,6:7454.
[28] Harden JL, Lewis SM, Pierson KC, et al. CARD14 expression in dermal endothelial cells in psoriasis[J]. PLoS One,2014,9(11):e111255.
[29] Sun Y, Zhang J, Zhou Z, et al. CCN1, a Pro-Inflammatory Factor, Aggravates Psoriasis Skin Lesions by Promoting Keratinocyte Activation[J]. J Invest Dermatol,2015,135(11):2666-2675.
[30] Li R, Wang J, Wang X, et al. Increased βTrCP are associated with imiquimod-induced psoriasis-like skin inflammation in mice via NF-κB signaling pathway[J]. Gene,2016,592(1):164-171.
[31] Bai J, Liu Z, Xu Z, et al. Epigenetic downregulation of SFRP4 contributes to epidermal hyperplasia in psoriasis[J]. J Immunol,2015,194(9):4185-4198.
[32] Lv F, Yu Y, Wang G, et al. Mechanisms by which the N-terminal 24 amino acids of the p55 regulatory subunit of phosphatidylinositol 3-kinase affect endotoxin-induced cytokine release in human keratinocytes[J]. Mol Med Rep,2015,11(5):3753-3759.
