微循环固相微萃取技术检测甲基苯丙胺合成过程中的特征物质
2018-06-28张文竞赵海清谢颜明
刘 帅,张文竞,李 虹,赵海清,谢颜明
(1.昆明医科大学法医学院,昆明 650500;2.毒品分析及禁毒技术公安部重点实验室,昆明 650228;3.临沧市公安局,云南 临沧 677000;4.普洱市公安局,云南 普洱 6650002)
云南毗邻世界毒源地 “金三角”,边境线长达4060公里,特殊的地理位置使其成为境外毒品流入中国的重要通道。随着全球毒品问题的持续泛滥,新型合成毒品缴获量已远远超过海洛因等传统毒品。由于我国是化工原料生产大国,云南又成为合成毒品原料非法走私出境的重要通道,因此在边境口岸严格落实毒品和制毒化学品“双向查缉”措施,开展新型毒品合成模式识别的研究,对预防和打击新型合成毒品违法犯罪,遏制新型毒品持续蔓延的势头,具有重要意义。
本文通过建立毒品甲基苯丙胺合成过程中的特征反应中间体、副产物和杂质的提取及检测方法,为分析新型毒品合成路径与易制毒化学品之间的关联性提供技术支撑。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器
Varian 3900/2100T气质联用仪。顶空微循环固相微萃取仪、咪唑交联树脂固相微萃取柱(吉林省兴科利民科技开发有限公司)。
1.1.2 试剂
pH 8.2磷酸缓冲液、乙酸乙酯、无水乙醇、乙腈(均为分析纯,由北京赛林格公司提供)。
1.1.3 实验样品来源
实验样品为云南省边境地区破获的毒品案件所缴获的毒品,由缅甸籍人员境外走私入境,总计11份样品,由云南省公安厅禁毒局提供。
1.2 条件参数
1.2.1 GC参数
色谱柱:DB-5,30 m×0.25 mm×1.0 μm;进样口温度:250 ℃,初温:50 ℃,程序升温10 ℃/min,终温:300 ℃,维持10 min,无分流进样1 μL。
1.2.2 GC-MS参数
色谱柱:DB-5MS,30 m×0.25 mm×1.0 μm;进样口温度:250 ℃,初温:50 ℃,程序升温5 ℃/min,终温:280 ℃,维持10 min,传输线温度280 ℃;离子源温度150 ℃,电离方式:EI;扫描范围:33~400(m/z);无分流进样1 μL,溶剂延迟:4 min,载气:He,流速:恒速1 mL/min。
1.2.3 顶空微循环固相微萃取仪参数
提取温度:110 ℃;气体流速:3~5 mL/min。
1.3 样品处理方法
萃取柱的活化:将萃取柱依次用3 mL乙腈和3 mL乙酸乙酯冲洗后密封保存。
称取甲基苯丙胺样品(100±5)mg溶于1 mL无水乙醇超声溶解,放入微循环顶空固相富集装置与吸附柱联接,在110 ℃加热循环提取30 min,自然冷却至室温,取出固相微萃取柱中的吸附质放入小试管内加入0.5 mL的乙酸乙酯,提取液供气相色谱/质谱(GC/MS)、气相色谱(GC)分析。
称取甲基苯丙胺样品(100±5)mg,放入10 mL具塞试管内,加入3 mL pH 8.2的磷酸缓冲液,超声溶解5 min,利用0.5 mL乙酸乙酯提取,提取液供GC/MS、GC分析。
2 结果与讨论
2.1 样品处理方法的优化
微循环固相微萃取与常规顶空固相微萃取方法比较:常规顶空固相微萃取是利用内置的吸附针对样品瓶上层空间的目的物进行富集达到分离的目的。这种方法有两点不足,一是加热温度低,导致样品中沸点较高的目的物在上层空间中浓度过低或根本就没有进入上层空间,无法对其进行富集和分离,如提高样品瓶加热温度,会导致低沸点化合物产生热脱附;二是受吸附容量限制,在竞争吸附过程中,不能被有效吸附,每提取一次只能进行一次分析。针对这两点不足,微循环顶空固相微萃取技术采用加热分离冷阱扑集方法,可以在较高温度下进行热分离,可以使沸点相差较大的物质同步进行热分离,固相微萃取柱的使用,使吸附容量的空间有了较大的提升,可以满足不同浓度目的物富集分离的需求,微溶剂解吸,可以保障一次样品前处理用以进行多次分析的需求。
2.1.1 固相微萃取柱内填充吸附材料的选择
常规固相微萃取柱是表面涂层为苯乙烯与硅酮交联树脂的固相微萃取纤维,此项研究中我们将固相微萃取柱制成带有不同涂层材料的微球进行使用,这样做的好处是提高吸附容量和结果的再现性。
比较苯乙烯与氮吡咯烷酮交联树脂、咪唑交联树脂、胺基树脂、硅藻土烧结微球四种涂层材料可知(见图1):涂层为胺基树脂与咪唑交联树脂的微球萃取目标杂质的种类较多;而苯乙烯与氮吡咯烷酮交联树脂萃取杂质干扰杂质较多;硅藻土烧结微球萃取效果较差。

图1 不同吸附材料提取的结果(a.苯乙烯与氮吡咯烷酮交联树脂;b.咪唑交联树脂;c.胺基树脂;d.硅澡土烧结微球)Fig.1 The effect of different adsorbents on the extraction
由于苯乙烯与氮吡咯烷酮交联树脂、胺基树脂是两种物质共聚形成,所以在萃取加热时可能产生降解,导致杂质峰过多,咪唑交联树脂是单体共聚形成,没有杂质峰出现,且其吸附率已满足鉴定需要,所以选择咪唑交联树脂做为固相微萃取吸附质。
2.1.2 提取温度
提取温度对提取效果有较大影响,温度过低会使高沸点物质在气相中所占比例降低,太高会导致甲基苯丙胺的相对含量升高影响分离效果,盐酸甲基苯丙胺的熔点约为130 ℃,所以提取温度应在80~120℃之间,选择90、110、120 ℃进行测试,结果表明,在90℃时部分杂质相对含量降低,120 ℃甲基苯丙胺相对含量急剧升高,导致其它杂质含量降低。因此110 ℃是最佳选择(图2)。

图2 不同温度提取的结果(a.120 ℃;b.90 ℃;c.110 ℃)Fig.2 The effect of different temperature on the extraction
2.1.3 关于吸附时间
比对15、30、60 min不同吸附时间的提取效果,最佳时间与文献报道的30 min[1]相同(图3)。
当前报道的样品处理方法主要是液/液萃取(LLE)、固相微萃取/热脱附(SPME/DT)法[2-4]。在LLE萃取甲基苯丙胺合成过程中的特征杂质时,大量主成分进入提取液,干扰待测微量或痕量的杂质目标物,同时造成毛细管色谱柱过载,影响分离效果;虽然SPME/DT在甲基苯丙胺合成模式识别方面的应用已有报道[5],但仍存在低通量、提取率低等不足,未能满足特征杂质鉴定时的多样品检测,而且,收缴的毒品中甲基苯丙胺是以盐酸盐的形式出现,与甲基苯丙胺相比其熔点、沸点有极大提高,其在气相中所占比例相对降低;因此我们采用微循环顶空固相富集/微溶剂脱附法对样品进行处理,不仅有效降低甲基苯丙胺的相对含量,同时还对其它痕量杂质起到富集作用。唯一不足的是,此方法会因溶剂峰和延迟采集丢失一部分低沸点的目标化合物,对所用溶剂的判别带来影响,目前尚无法解决,如在LLE中有大量乙酸被检出,而在微循环顶空固相富集/微溶剂脱附法中没能检出;但在相关性检验时会考虑毒品保存环境的影响,易挥发性物质不纳入数据处理范围,因此对鉴定结论不会产生影响,有文献报道只有在实验室自选合成的样品中才能检出乙醇、乙醚等易挥发性溶剂,而在收缴的样品中没能检出[5]。
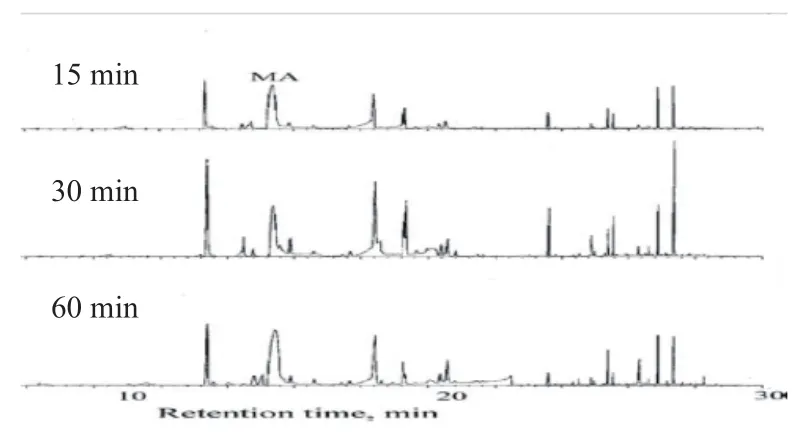
图3 不同吸附时间对提取效果的影响Fig.3 The effect of different adsorption time on the extraction
2.2 微循环顶空固相富集/微溶剂脱附法检测样品
采用微循环顶空固相富集/微溶剂脱附法检测境外走私入境的11份样品,分析其杂质成分。在对甲基苯丙胺合成方法进行研究中,所选目标化合物来自文献[6 -11]。目标化合物选择标准是:1)在收缴的毒品中出现有较高的概率;2)在溶液中有良好稳定性;3)在进行色谱分析时峰面积有良好的再现性;4)在色谱图中容易被鉴别;5)在本次实验中虽然出现频率较低或没有出现,但在多数文献中被引用。
由实验可知,待测样品与上述文献中共有的杂质为苯甲醛、苯胺、苄基氯、苯丙烯、反式1,2-二甲基-3-苯基环乙亚胺、苯基丙酮、苯丙胺、二甲基苯丙胺、N-乙基- N,π-二甲基苯乙胺、N-甲酰基甲基苯丙胺、乙酰甲基苯丙胺、N-乙酰麻黄碱、甲酰苯丙胺、3-(1-环己烯基)-3-乙烷基 -2,6-哌啶二酮;共有的特征物质为未知物1(基峰m/z58)、未知物2(基峰m/z120)、反式1,2-二甲基-3-苯基环乙亚胺、N-甲酰基甲基苯丙胺、乙酰甲基苯丙胺,见图4。

图4 样品中未知物的质谱图Fig.4 Mass spectrograms of the unknown substances in the sample
综上所述,本文建立的方法可用于缴获毒品甲基苯丙胺合成过程中的特征反应中间体、副产物和杂质的检测。
[1] KUWAYAMA K, TSUJIKAWA K H, KANAMORI T, et al.Identification of impurities and the statistical classification of methamphetamine using headspace solid phase microextraction and gas chromatography–mass spectrometry[J]. Forensic Science International, 2006,160(1): 44–52.
[2] ANDERSSON K, LOCK E , JALAVA K , et al. Development of a harmonised method for the prof i ling of amphetamines VIE valuation of methods for comparison of amphetamine[J]. Forensic Science International ,2007,169:86–99.
[3] KUWAYAMA K, INOUE H, KANAMORI T, et al. Contribution of thermal desorption and liquid–liquid extraction foridentification and profiling of impurities in methamphetamineby gas chromatography–mass spectrometry[J]. Forensic Science International, 2007,171(1):9–15.
[4] INOUE H, KANAMORI T, IWATA Y T, et al. Methamphetamine impurity prof i ling using a 0.32 mm i. d. nonpolarcapillary column, Forensic Science International. 2003,135 (1):42–47.
[5] KUWAYAMA K, TSUJIKAWA K, MIYAGUEHI H, et al. Identif i cation of impurities and the statistical classif i cation ofmethamphetamine using headspace solid phase microextraction and gas chromatography–mass spectrometry[J].Forensic Science International,2006,160(1): 44–52.
[6] REMBERG B, STEAD A H. Drug characterization/impurity prof i ling, with special focus on methamphetamine: recent work of the United Nations International Drug Control Programme[J].Bulletin on Narcotics. 1999,51(1):97–117.
[7] ANDERSSON K, LOCK E, JALAVA K, et al, Development of a harmonised method for the prof i ling of amphetamines VI Evaluation of methods for comparison of amphetamine[J]. Forensic Science International,2007,169(6): 86–99.
[8] SKINNER H F. Methamphetamine synthesis via hydriodic acid/red phosphorus reduction of ephedrine[J]. Forensic Science International,1990,48(2):123–134.
[9] WINDAHL K L, MCTIGUE M J, PEARSON J R, et al. Investigation of the impurities found in methamphetamine synthesised from pseudoephedrine by reduction with hydriodic acid and red phosphorus[J]. Forensic Science International,1995,76(2):97–114.
[10] INOUE H, KANAMORI T, IWATA Y T, et al, Methamphetamine impurity prof i ling using a 0.32 mm i. d. nonpolar capillary column[J].Forensic Science International,2003,135(1):42–47.
[11] PERSON E C, MEYER J A, VYVYAN J R, Structural determination of the principal byproduct of the lithium-ammonia reduction method of methamphetamine manufacture[J].Journal of Forensic Sciences, 2005,50 (1):1–9.
