顶空固相微萃取−气相色谱串联质谱法检测石油样品中的痕量多环芳烃
2018-06-28薛锦锋田琳琳梁丽军
薛锦锋,田琳琳,沈 磊,梁丽军
(浙江省嘉兴市公安局刑科所,浙江 嘉兴314001)
多环芳烃(polycyclic aromatic hydrocarbons, PAHs)是煤、石油、木材、烟草、有机高分子化合物等有机物不完全燃烧时产生的挥发性碳氢化合物,广泛分布于天然环境中,属于持久有机污染物,具有极强的致癌毒性。迄今已发现有200多种PAHs,有相当部分具有致癌性,其中常见的有16种同类物质[1]。石油是多环芳烃在自然界的主要存在源,在与石油相关的案件中,多环芳烃痕量组分的检测是石油检测的一个重点。近年来,顶空固相微萃取(HS−SPME)技术逐渐被人们关注,因其集取样、萃取及富集于一体,萃取快速、操作简单,便于实现自动化而广泛用于挥发性物质分析研究中。多采用固相微萃取-气质联用技术进行检测,但单级质谱技术的分析灵敏度较低,不能满足痕量组分的检测。本文在文献报道[2-4]基础上,利用顶空固相微萃取−气相色谱−三重四极杆串联质谱(GC/MS/MS)方法,对石油中的16种多环芳烃,采用多离子反应监测(MRM)技术,选择适当的母离子进行二次质谱分析,获得了比单级质谱选择离子检测(SIM)技术更多的碎片信息(母离子加子离子),同时有效去除了基质离子和其它物质的干扰,为石油样品中的多环芳烃痕量组分的检测提供了方法和依据。
1 材料与方法
1.1 仪器与试剂
Bruker SCION TQ−456GC 三重四极杆气相色谱/质谱联用仪及液体-顶空-固相微萃取三合一自动进样器(美国,布鲁克公司);固相微萃取装置:65 μm PDMS/DVB、100 μm PDMS 和 85 μm PA 3 种萃取头(美国,supelco公司)。
16种 PAHs混合标准溶液40 mg/L(美国,sigma-Aldrich公司);晕苯标准溶液0.5 mg/mL(美国,sigma-Aldrich公司)。尾油样品:原样稀释10倍,主要测定晕苯的含量;QLH样品:齐鲁尾油原样,稀释500倍,含烷烃与芳烃;A样品:QLH里萃取出的芳烃,稀释约50倍;PAHs混合标准溶液配制:取适当PAHs标准溶液与晕苯标准溶液混合,配制成浓度为1 mg/L的17种PAHs混合标准溶液(含内标物氘代对三联苯)。所用溶剂均为二氯乙烷。
1.2 实验条件
1.2.1 顶空固相微萃取条件
SPME萃取头(65 μm PDMS/DVB):首次使用前于300 ℃老化1 h;萃取方式:顶空自动;搅拌速度:500 r/min;样品预热平衡时间:20 min;样品平衡和萃取温度:40 ℃;解析温度:300 ℃;解析时间:100 s。
1.2.2 气相色谱条件
进样口温度:300 ℃,不分流进样;载气流速:1 mL/min;色谱柱:VF-5MS柱;柱温:柱起始温度为70 ℃,保持1 min,以25 ℃/min的速率升至140℃,后以10 ℃/min的速率升至 240 ℃,再以5 ℃/min 的速率升至300 ℃,保持7.2 min。
1.2.3 质谱条件
EI源;离子源温度:250 ℃;传输线温度:290 ℃;m/z 50-550;碰撞气:2 mTorr;溶剂延迟:4.5 min。
1.3 样品制备
准确移取3.00 mL待测样品于20 mL规格的顶空样品瓶中,压紧瓶盖放入自动进样盘中,固相微萃取借助自动进样器完成。
2 结果与讨论
2.1 质谱条件的优化
为获得最佳分析结果,保证对目标物定性定量的准确性,对待测物的母离子、产物离子、碰撞能量等质谱参数进行了优化。首先采用全扫描模式获得待测物的母离子,再用产物离子扫描模式通过优化碰撞能量获得产物离子,最后采用优化的质谱参数在MRM模式对待测物进行定性定量分析。经过优化,得到了较为理想的分析结果,优化后的质谱条件见表1。图1为1 mg/L PAHs混合标准溶液直接进样,全扫描测定得到的总离子流图。

表1 PAHs 的MRM质谱条件Table 1 MRM conditions for PAHs
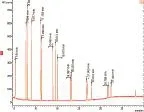
图1 1 mg/L PAHs混合标准溶液总离子流Fig.1 Total ion chromatograms of PAHs standard solution (1 mg/L)
2.2 顶空固相微萃取条件的优化
2.2.1 萃取纤维涂层的选择
本实验比较了常用的100 μm PDMS、65 μm PDMS/DVB(MW50-300)和 85 μm PA 3 种固相微萃取纤维涂层对PAHs的吸附效果,实验表明,萃取率大小 依 次 为 65 μm PDMS/DVB(MW50-300)>85 μm PA >100 μm PDMS。因此,本文选择 65 μm PDMS/DVB(MW50-300)作为萃取纤维涂层。
2.2.2 萃取温度和时间的优化
分别在30、40、50、60 ℃下进行萃取和检测,结果表明:随着萃取温度提高,萃取率有相应的提高,当温度超过40 ℃后,样品的萃取量反而减少。本实验选择40 ℃作为萃取温度。固定温度为40 ℃,对达到吸附平衡所需时间进行考察,改变萃取时间分别为10、20、30、40 min,结果表明:随着萃取时间增加,萃取率相应增加,20 min达到吸附平衡,因此选择萃取时间为20 min[5-7]。
2.3 线性方程、相关系数及灵敏度的检测
分别配制浓度为1、4、20、100、1000 μg/L的标准混合溶液,根据本文建立的方法进行检测。以定量离子的质谱峰面积为纵坐标(y),各目标物的含量为横坐标(x,μg/L),进行线性回归,用浓度0.1μg/L的PAHs混合标准溶液,以S/N=3确定检出限,以S/N=10确定定量限,结果见表2。
2.4 样品全扫描与MRM扫描结果比较
比较样品全扫描与MRM扫描结果可知,MRM扫描模式能够排除复杂石油样品的基质干扰,提高了检测的灵敏度和准确度,避免了假阳性结果的产生[8-9],结果见图 2~4。

表2 多环芳烃的线性方程、相关系数、检出限、定量限Table 2 Linear equation, correlation coeff i cient, LOD and LOQ of PHAs

图2 尾油样品的全扫描与MRM扫描TIC图Fig.2 Total ion chromatograms of tail oil with full and MRM scanning

图3 QLH尾油样品全扫描与MRM扫描TIC图Fig.3 Total ion chromatograms of QLH tail oil with full and MRM scanning

图4 样品A全扫描与MRM扫描TIC图Fig.4 Total ion chromatograms of sample A with full and MRM scanning
3 结论
本文采用顶空固相微萃取-气相色谱串联质谱方法,通过优化萃取条件和质谱条件,建立了一种痕量检测石油样品中的多环芳烃的简单有效的方法,检出限和定量限测定结果表明,方法的灵敏度高,各PAHs化合物在10~500 μg/L浓度范围内相关系数在0.999以上,线性关系良好。实验结果表明顶空固相微萃取-气相色谱串联质谱检测具有较高的排除基质干扰能力以及灵敏度优势,完全能满足案件中有关石油样品中的多环芳烃痕量组分的检测需求。
[1] 李煜婷,杜宇豪,李志,等. 自动固相萃取-高效液相色谱测定炼化废水中多环芳烃的方法研究[J].油气田环境保护,2016(5):40-43.
[2] 汪瑾彦,陈大舟,汤桦,等. 水体沉积物和土壤中多环芳烃的分析方法研究[J]. 现代仪器,2010,14(2):11-15.
[3] 梁柱,余雯静,孙欣阳. 加速溶剂萃取-色谱质谱联用法测定土壤中的多环芳烃[J]. 化学分析计量,2009,18(3):45-48.
[4] 邢若奎. 几种溶剂油燃烧残留物的ATD/GC/MS法检验[C]//第六届全国微量物证检验学术交流会论文汇编,2007:139-142.
[5] 李达,张琦,蔡璇. 顶空固相微萃取-气相色谱质谱联用技术分析桂油中香气成分[J]. 检验检疫学刊,2013(5):44-49.
[6] 陈旭东,周红. 固相微萃取-色谱/质谱技术分析水中汽油成分[J]. 理化检验:化学分册,2000,36(10):465-466.
[7] 梁丽军,何中央,张海琪,等. 固相萃取-高效液相色谱串联质谱法测定鱼类中地塞米松残留量[J]. 理化检验:化学分册,2009,45:1-4.
[8] 梁丽军,张海琪,何中央,等. 高效液相色谱-串联质谱法测定水产品中氢化可的松残留量[J]. 食品科技,2009,34(9):260-264.
[9] 魏万里. SPE-GC-MS/MS-SRM法快速检测尿液中12种常见毒品成分及代谢物[J]. 中国人民公安大学学报,2012(3):27-31.
