糖尿病肾病肾小管损伤机制
2018-06-27综述刘志红审校
李 申 综述 刘志红 审校
糖尿病肾病(DN)是糖尿病最常见的微血管并发症之一。随着我国糖尿病患病率不断增加,DN发病率也随之升高。据调查研究显示, DN已超越肾小球肾炎,成为我国住院患者慢性肾脏病(CKD)的首要病因[1]。美国近20年的调查研究数据显示,糖尿病的重要并发症(如心脑血管事件等)得到有效控制时,糖尿病导致终末期肾病(ESRD)的发生率并无明显降低[2],提示目前我们仍然缺乏有效干预手段延缓DN的进展。
DN病理改变包括肾小球硬化、肾小管间质纤维化及肾血管病变。目前DN的机制研究多集中于肾小球损伤,且临床上用于评判DN病情的指标也多基于肾小球结构和功能的改变[3],但肾小管间质占肾实质90%以上,肩负着多种重要功能,肾小管间质损伤在DN进展过程中发挥了重要作用。近年来,DN的肾小管间质损伤研究进展迅速,本文就相关进展进行总结。
糖尿病的肾小管结构和功能改变
肾单位是肾脏基本功能单元,由肾小球、肾小管及肾间质构成。肾小管包括近端肾小管和远端肾小管,其中近端肾小管通过包囊尿极直接与壁层上皮细胞相连。近端肾小管细胞管腔面附有刷状缘,可以增加细胞表面积,利于肾小管重吸收。此外,它是一种代谢旺盛的细胞,富有线粒体、溶酶体等细胞器[4],对能量需求大,其对损伤非常敏感。
糖尿病时,肾小管管腔中高浓度的葡萄糖和次级代谢产物,可诱导肾小管上皮细胞损伤。经肾小球滤过的葡萄糖有99%被近端肾小管重吸收,特别是近端肾小管S1区段中的葡萄糖转运蛋白2(Sodium-glucose Cotransporter-2,SGLT2),介导了肾脏中90%葡萄糖的重吸收[5]。研究表明,2型糖尿病(T2DM)患者,即使血糖控制良好,其肾糖阈及葡萄糖重吸收能力(maximum renal glucose reabsorptive Capacity,TmG)也高于正常人群[6]。近端肾小管上皮细胞在摄入超量葡萄糖后,可诱导多种细胞因子的表达和释放,如血小板衍生生长因子、胰岛素样生长因子1和表皮生长因子(EGF)等,继而引起肾小管上皮细胞增殖和肥大[7]。高糖状态还会导致肾小管上皮细胞线粒体功能障碍,从而释放更多氧化应激产物,介导细胞损伤。此外,肾小管在重吸收葡萄糖的同时,亦可通过SGLT2等导致Na+重吸收的增加,通过致密斑的管球反馈机制,导致肾小球出现高灌注、高压力和高滤过的变化,加重肾脏损伤。因此,临床应用SGLT2抑制剂,如达格列净,增加尿糖排泄,从而控制血糖,保护肾功能[6]。
此外,有研究发现肾小管结构异常在DN早期即出现,甚至早于肾小球结构异常,且部分患者肾小球结构没有显著异常就可出现蛋白尿等临床表型。近端肾小管的结构特征使得其在糖尿病状态下更容易受到打击。当包囊和近端肾小管的连接处受到损伤而发生萎缩,会形成无功能的无管肾小球。Najafian等[8-9]发现,即使在估算的肾小球滤过率(eGFR)正常或中度受损的DN患者中,就有17%肾小球为无管肾小球,51%肾小球附着于萎缩的肾小管。White等[10]也发现,在T2DM合并DN患者中,有7%患者存在无管肾小球,即使是在微量白蛋白尿的DN患者中,仍有26%患者出现球管连接异常,其异常比例与肌酐清除率负相关(r=-0.70,P=0.011)。
代谢异常加重肾小管损伤
肾小管细胞是线粒体富集细胞,需要大量三磷酸腺苷(ATP)维持正常功能。糖尿病状态下,各种代谢底物传递紊乱导致ATP合成异常,从而影响肾小管上皮细胞正常功能,也是启动肾小管损伤和间质纤维化的始动因素之一。
高血糖可通过多种葡萄糖代谢途径诱导代谢异常,并诱导线粒体功能障碍,线粒体分裂增多,过度产生活性氧。Sun等[11]发现高血糖状态下,肾小管上皮细胞线粒体的形态和功能发生改变,线粒体中Ras相关蛋白1b(Ras-related protein 1,Rap1b)的GTP酶活性和Bcl-2的表达下调,Bax表达上调,导致细胞凋亡增加。过表达Rap1b可改善高糖诱导的线粒体线形态与功能障碍,减轻细胞凋亡。近期Qi等[12]分析了糖尿病患者与DN患者的肾小球转录组数据,发现相比于健康人群以及DN患者,丙酮酸激酶M2(Pyruvate kinase M2,PKM2)在未并发DN的糖尿病患者中显著上调。PKM2是糖酵解的限速酶之一,在糖酵解的产能中发挥重要作用,可以使磷酸烯醇式丙酮酸和ADP变为ATP和丙酮酸。进一步的机制研究证实,PKM2活化可增加糖酵解,抑制毒性葡萄糖代谢物的产生,诱导线粒体融合,从而恢复线粒体功能来防止DN,这也证实了PKM2在未来作为干预DN发生发展的分子靶点的价值。
除了高血糖外,糖尿病状态下脂肪酸代谢紊乱也可导致肾小管间质损伤。正常情况下,肾小管上皮细胞优先通过脂肪酸氧化(FAO)来提供能量,即使在疾病状态导致脂肪酸代谢下降时,其依旧优先利用脂肪酸供能,而非通过燃烧葡萄糖提供能量。有研究发现,与正常对照相比,肾小管间质纤维化的肾脏病患者和小鼠模型的肾小管上皮细胞内的FAO的关键酶和调节因子的表达显著降低[13]。该研究发现, 抑制肾小管上皮细胞中的FAO,肾小管上皮细胞会出现ATP消耗增加、细胞死亡、细胞去分化和细胞内脂质沉积等纤维化表型,导致肾小管间质纤维化的发生。相反,恢复FAO可发挥减缓肾脏纤维化过程的作用。此外,很多microRNA也被发现可通过调节代谢途径,发挥影响DN发生与发展的作用。如miR-21可通过下调过氧化物酶体增殖物激活受体α加重肾小管损伤和纤维化[14]。
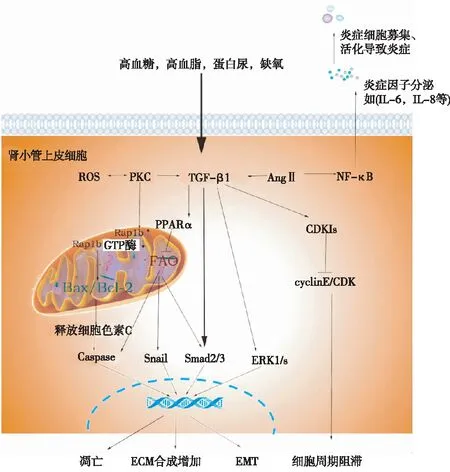
图1 肾小管上皮细胞在DN间质纤维化过程中的作用DN时,肾小管上皮细胞通过多种细胞途径诱发细胞凋亡,基质沉积,上皮间充质转分化,细胞周期阻滞及炎症等,从而导致间质纤维化;DN:糖尿病肾病;ROS:活性氧;PKC:蛋白激酶C;TGF-β1:转录生长因子β1;AngⅡ:血管紧张素Ⅱ;NF-κB:核因子κB;Rap1b:Ras相关蛋白1b;Caspase:含半胱氨酸的天冬氨酸蛋白水解酶;PPARα:过氧化物酶体增殖物激活受体α;FAO:脂肪酸氧化;ERK1/2:细胞外调节蛋白激酶1/2;CDKIs:细胞周期蛋白依赖性蛋白激酶抑制因子;cyclinE:细胞周期蛋白E;CDK:细胞周期蛋白依赖性蛋白激酶
蛋白尿加重肾小管损伤
过量尿蛋白对肾小管具有毒性作用。肾小管上皮细胞可通过megalin,cublin重吸收尿蛋白。暴露于高浓度白蛋白的近端肾小管上皮细胞会分泌趋化因子和炎性因子,如白细胞介素1β(IL-1β)、IL-6、IL-8、趋化因子配体2等[15],激活核因子κB(NF-κB)途径,加重肾脏炎症,导致肾脏损伤。此外,Wu等[16]发现,白蛋白超载还会使得肾小管上皮细胞出现上皮间充质转分化(EMT),诱导肾脏纤维化。并且越来越多研究发现,肾小管的蛋白超载可通过多种信号途径,如线粒体相关凋亡途径,内质网应激相关凋亡途径等导致肾小管上皮细胞凋亡。Erkan等[17]发现白蛋白可通过诱导线粒体释放细胞色素C,活化含半胱氨酸的天冬氨酸蛋白水解酶3和9,从而诱导肾小管上皮细胞凋亡(图1)。
肾小管损伤加重肾脏损伤
肾小球肾小管的损伤也可诱导肾小球的损伤。Hasegawa等[18]构建了肾小管特异性过表达Sirt1基因的链脲佐菌素(STZ)小鼠,发现可以显著减轻STZ小鼠蛋白尿。进一步的机制研究发现, Sirt1可调控肾小管上皮细胞烟酰胺单核苷酸(NMN)的分泌,而NMN可被逆向运送至肾小球,并被足细胞吸收,调控足细胞中紧密连接蛋白表达,从而影响肾小球功能。Grgic 等[19]利用白喉毒素细胞剔除系统,于小鼠肾小管上皮细胞特异表达白喉毒素受体,随后给予DT反复刺激,以单纯损伤近端肾小管的S1 /S2 段,但可以观察到肾小球硬化的发生。
肾血管DN发生过程中,在肾小球及小管间质发生形态改变之前,肾脏血流动力学就会发生变化,如血管活性物质失衡,出球小动脉收缩,管周毛细血管血流灌注降低等。这些变化会继而诱发肾组织慢性缺氧,导致或加重肾小管间质损伤。与此同时,肾小管上皮细胞的损伤也会导致肾血管的病变。体外培养近端肾小管上皮细胞表明,高糖环境可刺激其血管紧张素Ⅱ(Ang Ⅱ)的高表达。而Ang Ⅱ可直接损伤内皮细胞。有研究发现,Ang Ⅱ可导致肾脏管周毛细血管网密度降低,给予血管紧张素转换酶抑制剂(ACEI)则可缓解或逆转这一损伤[20]。
肾间质纤维化既往研究多认为CKD的始发因素是肾小球损伤,而肾小管损伤则继发于肾小球损害。近来,越来越多研究表明,肾小管损伤可能才是肾脏疾病的始动因素。一个有力的证据就是,在急性肾损伤(AKI)后,肾脏出现肾小管萎缩和间质纤维化等病理改变,但肾小球形态正常。有研究表明,AKI后,部分肾小管上皮细胞出现生长停滞和再分化失败,这类异常的肾小管上皮细胞可以表达促纤维肽,并且激活多个促纤维化信号通路,从而导致肾间质纤维化和CKD的发生[21]。
另一个证据来源于目前对早期DN患者微量白蛋白尿的研究。既往关于蛋白尿的研究都聚焦于肾小球的滤过能力,但由于近端肾小管上皮细胞具备蛋白重吸收的能力,因此DN患者的微量白蛋白尿是由于肾小球滤过障碍还是近端肾小管重吸收功能减退所致,仍存在争议。Russo等[22]选取12周STZ大鼠作为早期DN模型,用双光子显微镜观测了其体内白蛋白滤过及重吸收过程。结果发现,即使STZ大鼠和正常对照的蛋白尿水平不同,但是两组间肾小球滤过功能并无明显差异。而与正常对照相比,STZ组大鼠近端肾小管重吸收能力明显减弱,导致荧光标记的蛋白出现在终尿中,这提示早期观察到的蛋白尿是肾小管重吸收功能障碍所导致的。
肾小管损伤除诱发DN之外,还密切参与肾间质纤维化。在糖尿病情况下,近端肾小管及肾间质会发生一系列的结构性变化,如肾小管萎缩、间质性纤维化和管周毛细血管减少,其均与肾脏功能下降密切相关。肾小管间质纤维化更是多种CKD进行性发展的病理学基础。An等[23]通过对DN患者的肾组织切片染色分析发现,间质纤维化及肾小管萎缩程度与肾脏结局密切相关。此外,机制研究亦表明,DN时,肾小管上皮细胞转录生长因子β1表达增加,Wnt-β catenin等信号通路激活,多种信号通路之间交互作用,继而诱导细胞周期阻滞,凋亡,EMT,以及通过Smad家族增加促纤维化分子的转录等方式参与肾小管间质纤维化(图1)。
此外,基于肾小管损伤的肾脏病预后标志物的发现,也进一步证实了肾小管间质损伤在肾脏疾病进展中的重要作用。肝脏型脂肪酸结合蛋白、嗜中性粒细胞明胶酶相关的脂质运载蛋白、肾损伤分子 1等均可用于AKI的早期诊断。在DN人群中,上述生物标志物亦在微量白蛋白患者中升高,并且与eGFR下降相关,但这种相关性在经过蛋白尿等校正后,不再具有统计学意义[24]。Ju等[25]通过肾小管间质组织发现EGF在CKD患者与正常对照之间差异表达,并发现无论在尿液蛋白水平还是肾组织mRNA水平,EGF均与eGFR及eGFR下降斜率显著相关,有效预测CKD患者的预后,且针对其中的DN患者亚组分析表明,EGF是DN有效的预后生物标志物。
小结:肾小管损伤是DN发生发展中的关键一环。介导肾小管损伤的机制复杂,包括了代谢机制、血流动力学机制、免疫炎症机制、肾小管细胞同其他肾脏固有细胞的相互作用等。而肾小管损伤后,又可通过多种途径介导肾小球硬化和肾脏纤维化的过程。DN肾小管损伤机制研究在DN分子机制研究中占据重要地位,特别是目前一些新的研究方法,如组学研究、双光子显微镜、分子示踪技术等在肾小管损伤机制研究中的应用,有望揭示新的延缓DN疾病进展的分子干预靶点和手段,对于改善DN患者远期预后具有重要作用。
1 Zhang L,Long J,Jiang W,et al.Trends in Chronic Kidney Disease in China[J].N Engl J Med,2016,375(9):905-906.
2 Gregg EW,Li Y,Wang J,et al.Changes in diabetes-related complications in the United States,1990-2010.N Engl J Med,2014,370(16):1514-1523.
3 Gilbert RE.Proximal Tubulopathy:Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease.Diabetes,2017,66(4):791-800.
4 朱加明.肾脏解剖∥黎磊石,刘志红.中国肾脏病学.北京:人民军医出版社,2008:13-14.
5 Bakris GL,Fonseca VA,Sharma K,et al.Renal sodium-glucose transport:role in diabetes mellitus and potential clinical implications.Kidney Int,2009,75(12):1272-1277.
6 DeFronzo RA,Norton L,Abdul-Ghani M.Renal,metabolic and cardiovascular considerations of SGLT2 inhibition.Nat Rev Nephrol,2017,13(1):11-26.
7 Vallon V,Thomson SC.Renal function in diabetic disease models:the tubular system in the pathophysiology of the diabetic kidney.Annu Rev Physiol,2012,74:351-375.
8 Najafian B,Kim Y,Crosson JT,et al.Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy.J Am Soc Nephrol,2003,14(4):908-917.
9 Najafian B,Crosson JT,Kim Y,et al.Glomerulotubular junction abnormalities are associated with proteinuria in type 1 diabetes.J Am Soc Nephrol,2006,17(4 Suppl 2):S53-60.
10 White KE,Marshall SM,Bilous RW.Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy.Nephrol Dial Transplant,2008,23(11):3539-3545.
11 Sun L,Xie P,Wada J,et al.Rap1b GTPase ameliorates glucose-induced mitochondrial dysfunction.J Am Soc Nephrol,2008,19(12):2293-2301.
12 Qi W,Keenan HA,Li Q,et al.Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction.Nat Med,2017,23(6):753-762.
13 Kang HM,Ahn SH,Choi P,et al.Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development.Nat Med,2015,21(1):37-46.
14 Chau BN,Xin C,Hartner J,et al.MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways.Sci Transl Med,2012,4(121):121ra18.
15 Tang S,Leung JC,Abe K,et al.Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo.J Clin Invest,2003,111(4):515-527.
16 Wu HJ,Yiu WH,Li RX,et al.Mesenchymal stem cells modulate albumin-induced renal tubular inflammation and fibrosis.PLoS One,2014,9(3):e90883.
17 Erkan E,Devarajan P,Schwartz GJ.Mitochondria are the major targets in albumin-induced apoptosis in proximal tubule cells.J Am Soc Nephrol,2007,18(4):1199-1208.
18 Hasegawa K,Wakino S,Simic P,et al.Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes.Nat Med,2013,19(11):1496-1504.
19 Grgic I,Campanholle G,Bijol V,et al.Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis.Kidney Int,2012,82(2):172-183.
20 Lombardi D,Gordon KL,Polinsky P,et al.Salt-sensitive hypertension develops after short-term exposure to Angiotensin Ⅱ.Hypertension,1999,33(4):1013-1019.
21 Venkatachalam MA,Weinberg JM,Kriz W,et al.Failed Tubule Recovery,AKI-CKD Transition,and Kidney Disease Progression.J Am Soc Nephrol,2015,26(8):1765-1776.
22 Russo LM,Sandoval RM,Campos SB,et al.Impaired tubular uptake explains albuminuria in early diabetic nephropathy.J Am Soc Nephrol,2009,20(3):489-494.
23 An Y,Xu F,Le W,et al.Renal histologic changes and the outcome in patients with diabetic nephropathy.Nephrol Dial Transplant,2015,30(2):257-266.
24 Nielsen SE,Andersen S,Zdunek D,et al.Tubular markers do not predict the decline in glomerular filtration rate in type 1 diabetic patients with overt nephropathy.Kidney Int,2011,79(10):1113-1118.
25 Ju W,Nair V,Smith S,et al.Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker.Sci Transl Med,2015,7(316):316ra193.
