克隆整合对遮荫白夹竹分株根际土壤细菌生物特征的影响
2018-06-22陈劲松宋会兴
薛 阁,李 洋,陈劲松,宋会兴,*
1 四川农业大学风景园林学院, 成都 611130 2 四川师范大学生命科学学院, 成都 610101
由于克隆生长,同一克隆片段内的分株通过匍匐茎、根状茎等水平结构连接在一起,并且相连分株间存在物质传递(如光合产物、水分、养分等),这被称为克隆整合(clonal integration)[1]。对光合产物、矿质养分、水分等物质的克隆整合格局同位素示踪以及染料标记等的研究结果表明,同化产物在克隆片段内既可以顶向传输(acropetal transport),即流向远端分株(distal ramet);也可以基向传输(basipetal transport),即流向近端分株(proximal ramet)[2]。因此,克隆整合可能是克隆植物维持生态优势的重要手段,其通过分株间生理整合缓解资源异质性(光照等)带来的压力。
释放到根际环境中的植物根系分泌物被认为是投入到土壤中易分解碳最主要的来源[3]。这些易分解的有机质成为异养根际微生物重要的能量来源、结构物质,能够促进根系微生物的快速生长和繁殖[4]。土壤有机质矿化作用是受土壤酶催化的复杂生物化学过程,这其中土壤酶主要来源于土壤微生物[5-6]。土壤微生物代谢活动产生的酶类,一部分参与自身生理代谢,另一部分通过排泄或分泌到土壤基质中,被认为是为土壤微生物活性的敏感指示[7]。因此,根际生态过程(如有机质降解等)及养分有效性可能受到土壤微生物活性及其群落结构变化的影响。
土壤是一个复杂的、动态变化的系统,关于土壤代谢过程(有机质降解、硝化过程、土壤呼吸等)以及特定土壤酶活性(脲酶、蛋白酶、木质素酶等)的研究,间接揭示了土壤微生物在生态系统养分循环中的重要作用[8]。土壤微生物群落结构主要由群落的种类和种间差异来描述。土壤微生物通常种类繁多、数量巨大,加之土壤介质本身的复杂多变,传统的分离培养方法仅仅能筛选出极少数的土壤微生物(1%—10%),而绝大多数土壤微生物在实验室条件下难以培养、鉴定[9]。20世纪90年代以来,DNA测序技术快速发展并在分子生态学上得到广泛应用,16S rDNA测序被越来越多的研究人员应用于环境中细菌的群落结构分析[10-11]。本研究中,我们采集白夹竹遮荫分株(近端分株,远端分株)的根际土壤,对溶解性有机碳、微生物生物量、土壤酶活性进行测定以研究克隆整合对异质性光照下白夹竹分株根际土壤生物过程的影响;同时,提取根际土壤基因组DNA,进行16S rDNA测序来研究分株间资源共享对异质性光照下白夹竹分株根际土壤的细菌群落结构的影响。
1 材料与方法
1.1 实验材料
白夹竹(Phyllostachysbissetii)又称蓉城竹、四川刚竹,是禾本科(Gramineae)刚竹属(Phyllostachys)小型竹种,竿高3—6m,粗约2cm,它通过地下根状茎(竹鞭,rhizome)在水平空间拓展,是典型的克隆植物。产浙江、四川,竿作柄材或篾用,笋食用,同时是大熊猫的主食竹之一。
1.2 实验设计
实验样地位于四川省邛崃市南宝山镇(103°14′14″E, 30°14′31″N),海拔1217m,年均温16.3℃,年降雨量1117.3mm。2015年12月,选择地势较为平缓的白夹竹纯林作为实验处理区域。选择分株大小、竹鞭间距一致的白夹竹克隆片段(包含通过竹鞭相连的白夹竹分株),分别标记近端分株和远端分株。
整个实验包含两个处理组:顶向处理组,即克隆片段中远端分株处于80%遮荫状态,近端分株处于正常光照;基向处理组,即克隆片段中近端分株处于80%遮荫状态,远端分株处于正常光照。分株间的根状茎处于割断或连接处理。实验过程采取如下处理,以排除外界土壤对遮荫分株根际生态过程的影响:以白夹竹分株竿基部为中心(50cm×50cm)进行挖沟,深度50cm,之后对分株根部土块周围包裹聚乙烯膜和PVC板材。聚乙烯膜和PVC板材主要用于防止外部土壤环境中水分、养分渗入到白夹竹分株根部土块中。同时,分株根部土块表面覆盖遮阳网,以防止落叶等凋落物对实验的影响。实验设计如图1所示。每种实验处理进行10次重复。上述实验处理工作于2015年1月中旬全部完成。
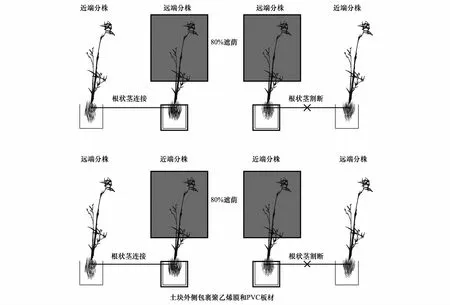
图1 实验设计图Fig.1 Schematic diagram of experiment design
2016年8月12日,采用“抖根法”取得克隆片段内进行遮荫处理的白夹竹分株根际土壤[12-13],采集附着于根系表面约4mm以内的土壤作为根际土壤,并手工挑出其中的植物残体,过筛(<2mm)后储存于干燥灭菌的自封袋中,并保存于-80℃超低温冰箱中备用。
1.3 土壤理化指标测定
根际土壤微生物生物量碳、氮(MBC or MBN)的测定采用氯仿熏蒸法(chloroform-fumigation extraction method with fumigation at atmospheric pressure, CFAP)[14]。用0.5mol/L K2SO4溶液对进行氯仿熏蒸处理以及未进行熏蒸处理的根际土壤进行浸提,浸提液过滤后通过TOC/TN分析仪(TOC-L analyzer, SHIMADZU, Japan)进行溶解性有机碳(DOC)以及溶解性有机氮(DON)含量的测定。微生物生物量的计算通过以下的公式进行计算[15]。
MBC(N)=2.22×EB
式中,EB是进行氯仿熏蒸处理与未进行熏蒸处理的浸提液溶解性碳、氮含量差值。
N-乙酰基-β-D-氨基葡萄糖酶(N-acetyl-β-D-glucosaminidase,NAGase)活性采用Parham和Deng[16]的方法进行测定,NAGase活性表达为每克鲜土培养1h后释放的ρ-nitrophenol的微克数;脲酶(Urease)活性的测定根据Kandeler等[17]的方法,以尿素为底物,脲酶活性表达为每克干土在37℃下培养2h后每毫升浸提液中水解氮的微克数。
1.4 土壤样本高通量测序
采用“GENEOUT土壤DNA提取试剂盒”(LabGene,成都),按照操作说明提取土壤微生物基因组DNA,并进行1%凝胶电泳检测。引物338F(5′-ACTCCTACGGGAGGCAGCAG- 3′)和806R(5′-GGACTA CHVGGGTWTCTAAT- 3′)被用来扩增细菌16S rRNA基因的V3、V4可变区[18]。PCR反应采用20μL反应体系:4μL 5× Fast Pfu buffer溶液, 2μL 2.5 mmol/L dNTPs溶液, 0.4μL上游引物(5μmol/L)及0.4μL下游引物 (5μmol/L), 0.4μL Fast Pfu polymerase酶, 以及 10ng 模板DNA,补足ddH2O至20μL。PCR反应退火温度是55℃。进行PCR反应的特异性引物需要包含以下特征:1)合适的Illumina接头序列使得amplicons与流动槽(flow cell)连接;2)一个8bp的标签序列(index sequence),如barcode;3)特异性扩增上述目的片段[19]。同时为满足后续数据分析的准确性以及可靠性,PCR反应尽可能采用少的循环数,并保证每个样本扩增的循环数一致。
每个样品PCR反应重复3次,将同一样本的PCR产物混合后用2%的琼脂糖凝胶电泳检测。Amplicons采用Axy Prep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA)试剂盒进行纯化。扩增子用QuantiFluorTM-ST蓝色荧光定量系统(Promega,USA)进行检测定量,纯化后的扩增产物进行等摩尔量混合。PhiX Contral library与Amplicons library进行混合,然后在Illumina Miseq platform (Majorbio, Shanghai, China)进行双端测序(paired-end sequence,2×300)。
1.5 数据与生物信息分析
采用单因素方差分析(one-way ANOVA)分析根状茎连接与割断处理对遮荫白夹竹分株根际土壤酶活性、DOC和微生物生物量的影响。数据分析以及图表处理采用SPSS 22 软件(SPSS, Chicago IL USA) 和Origin Pro 9软件 (OriginLab Corp., Massachusetts USA)完成。
Miseq原始测序数据,根据标签序列将双端测序序列提取出来,进一步的read过滤处理包括:1)剔除引物序列;2)过滤read尾部质量值20以下的碱基,设置50bp大小的滑动窗口,若窗口内平均质量低于20从窗口开始截去后端碱基;3)移除小于50bp的truncated reads[20]。经过剪切,去除掉存在引物错配、不确定碱基的序列;同时,根据PE reads之间的重叠关系,将成对的reads拼接成一条序列,重叠序列的最小长度为10bp;拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列;根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2。经过质量控制、过滤的Contigs采用QIIME(version 1.17)软件进行后续剪切、去复和分类学归类。对于优化后的contigs提取非重复序列,主要为了降低分析中间过程冗余计算量;去掉没有重复的单序列。按照97%的相似性将非重复序列序列进行同源比对并聚类成可操作分类单元(OTU),聚类过程中通过UCHIME确认并剔除序列嵌合体,从而得到OTU代表序列;然后将所有优化后的contigs映射到OTU代表序列,选出与OTU代表序列相似性在97%以上的,归类到相应的OTU组别里[21]。然后,与数据库Silva Release119(https://www.arb-silva.de/documentation/release- 119)进行比对(uclust,identity 0.9)。
采用Mothur (v1.12.1)软件制作稀释性曲线以确定测序数据深度以及合理性,并计算反应菌群多样性的香农指数(Shannon index)。基于样本OTUs的主成分分析(PCA)由R语言完成,并Vegan 2.0 package作PCA图。
2 研究结果
2.1 土壤性质
顶向处理组中,根状茎连接的遮荫远端分株具有比根状茎切断遮荫分株更高的根际土壤DOC、MBC、MBN含量,其中MBC、MBN含量差异显著(图2);在基向处理组中,根状茎连接的近端分株遮荫后具有比根状茎割断处理的遮荫分株更高的DOC、MBC和MBN含量,差异均达到显著水平(图2)。
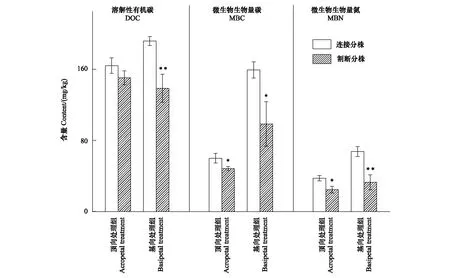
图2 遮荫分株根际土壤溶解性有机碳(DOC),微生物生物量碳(MBC),微生物生物量氮(MBN)含量Fig.2 Contents of DOC, MBC, MBN in the rhizosphere soil of shaded distal, proximal ramets差异显著性依次为** P<0.01, * P<0.05
无论在顶向处理组还是基向处理组中,根状茎连接的遮荫分株较根状茎切断的遮荫分株具有更高的NAGase、Urease活性(图3)。顶向处理组中,根状茎连接状态下的遮荫远端分株根际土壤的NAGase活性显著高于根状茎割断处理的遮荫远端分株,但这种显著性差异并没有在基向处理组中观测到;而对Urease活性来说,不论是顶向处理还是基向处理,差异均达到显著性水平(图3)。
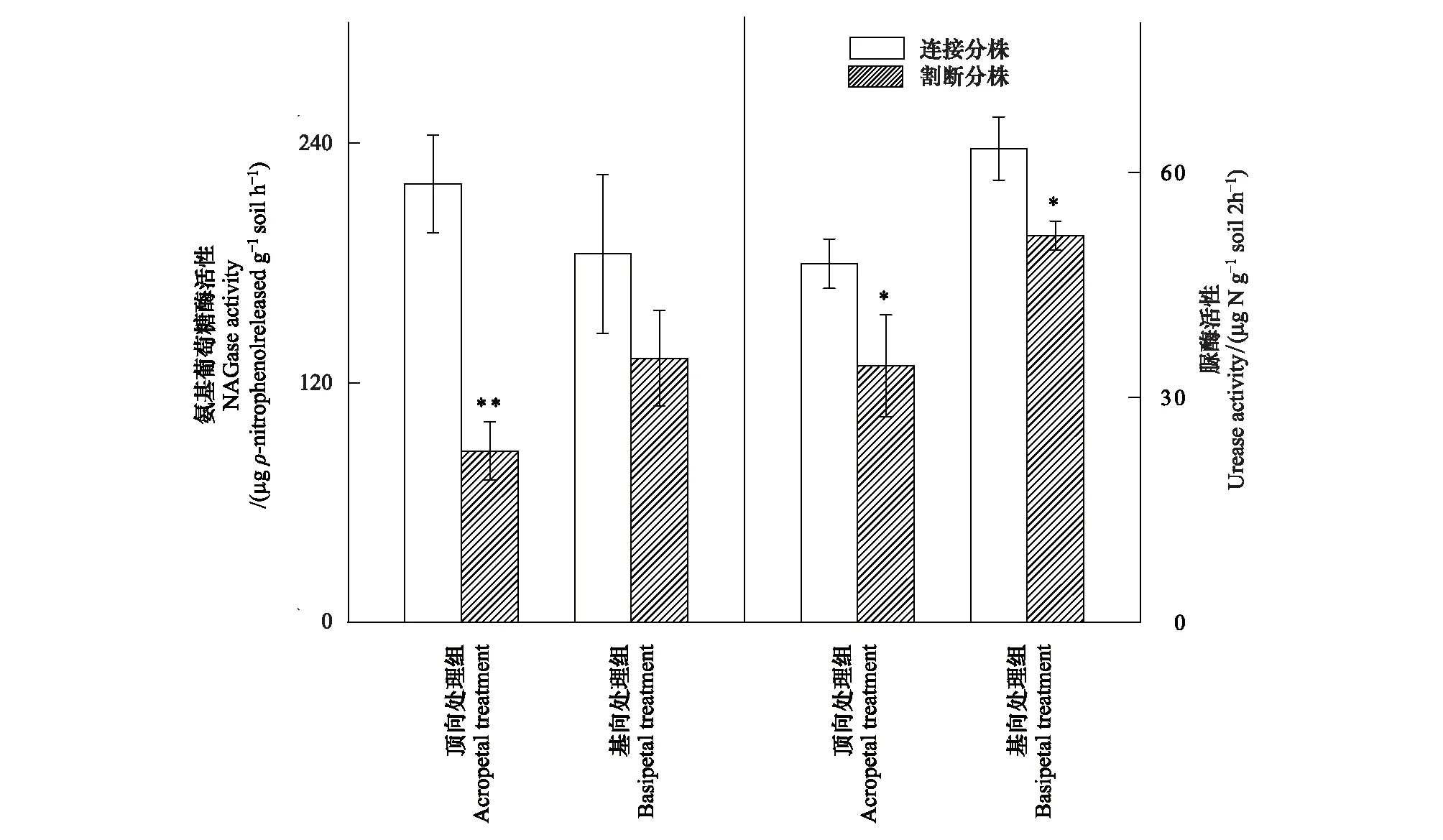
图3 遮荫分株根际土壤胞外酶(NAGase, Urease)活性Fig.3 Extracellular enzyme activities in the rhizosphere soil of shaded ramet
根状茎连接状态、同化产物传输方向以及二者交互作用对遮荫分株根际土壤特征的双因素方差分析结果如表1所示。根状茎连接状态(连接、断开)对遮荫分株根际土壤DOC、微生物生物量、胞外酶活性均产生了显著性影响;遮荫分株根际土壤微生物生物量、Urease活性显著则受到同化产物传输方向(顶向、基向)的显著影响;除Urease活性,根际土壤DOC、微生物生物量、NAGase活性均受到根状茎连接状态、同化产物传输方向以及二者交互作用的显著影响。
表1根状茎连接状态(连接、割断)、同化产物传输方向(顶向、基向)对遮荫分株根际土壤特征、土壤胞外酶活性的双因素方差分析结果
Table1Resultofsignificancetestforsoilpropertiesandsoilenzymeactivitiesintherhizospheresoilofshaded,distalorproximalramet.Theeffectofrhizomeconnectionstatus(connected or severed),directionofphotosynthatestransport(acropetal transport or basipetal transport)andtheirinteractionweretestedintwo-wayANOVAs
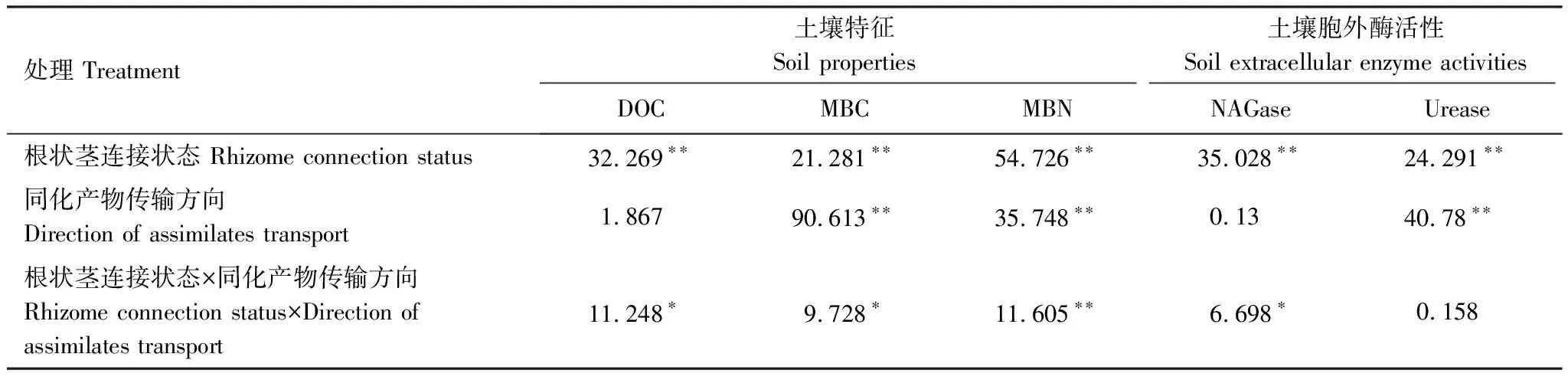
处理 Treatment土壤特征 Soil properties土壤胞外酶活性 Soil extracellular enzyme activitiesDOCMBCMBNNAGaseUrease根状茎连接状态 Rhizome connection status32.269∗∗21.281∗∗54.726∗∗35.028∗∗24.291∗∗同化产物传输方向Direction of assimilates transport1.86790.613∗∗35.748∗∗0.1340.78∗∗根状茎连接状态×同化产物传输方向 Rhizome connection status×Direction of assimilates transport11.248∗9.728∗11.605∗∗6.698∗0.158
表中数字为F值,显著性检验表示为 **P<0.01,*P<0.05; DOC:溶解性有机碳,dissolved organic carbon; MBC:微生物生物量碳,microbial biomass carbon;MBN:微生物生物量氮,microbial biomass nitrogen
2.2 Illumina Miseq测序分析
通过Illumina Miseq测序,从4个处理各3个重复共12个土壤样品中总共得到了556519条序列和18898条可操作分类单元(operational taxonomic unit, OTU)。样本的DNA文库中包含38478到55004个不等的序列(reads),OTU数目从1244到1755不等(表2)。稀释性曲线达到平台期表明测序序列的数据量是合理的,意即样本产生再多数量的序列也对最终不同样本中的OTUs数目没有太大贡献[22]。本研究中的稀释性曲线表明,根状茎连接的白夹竹遮荫近端分株根际土壤(Sample 2)中的OTUs数量与其余样品之间存在较大差异;同时样品4,8,9,3,11与样本1,12,5,6,7,10之间OTUs数目存在微弱差异。但整体来看,所有的样本的OTUs数目差距不大(图4,表2),表明不同根状茎连接状态(割断,连接)的白夹竹遮荫分株(远端分株,近端分株)根际土壤的OTU数目并不存在显著性差异。通过计算α多样性指数(Shannon index),发现克隆整合对白夹竹遮荫分株(远端分株,近端分株)根际土壤细菌群落多样性的影响并没有达到显著水平。
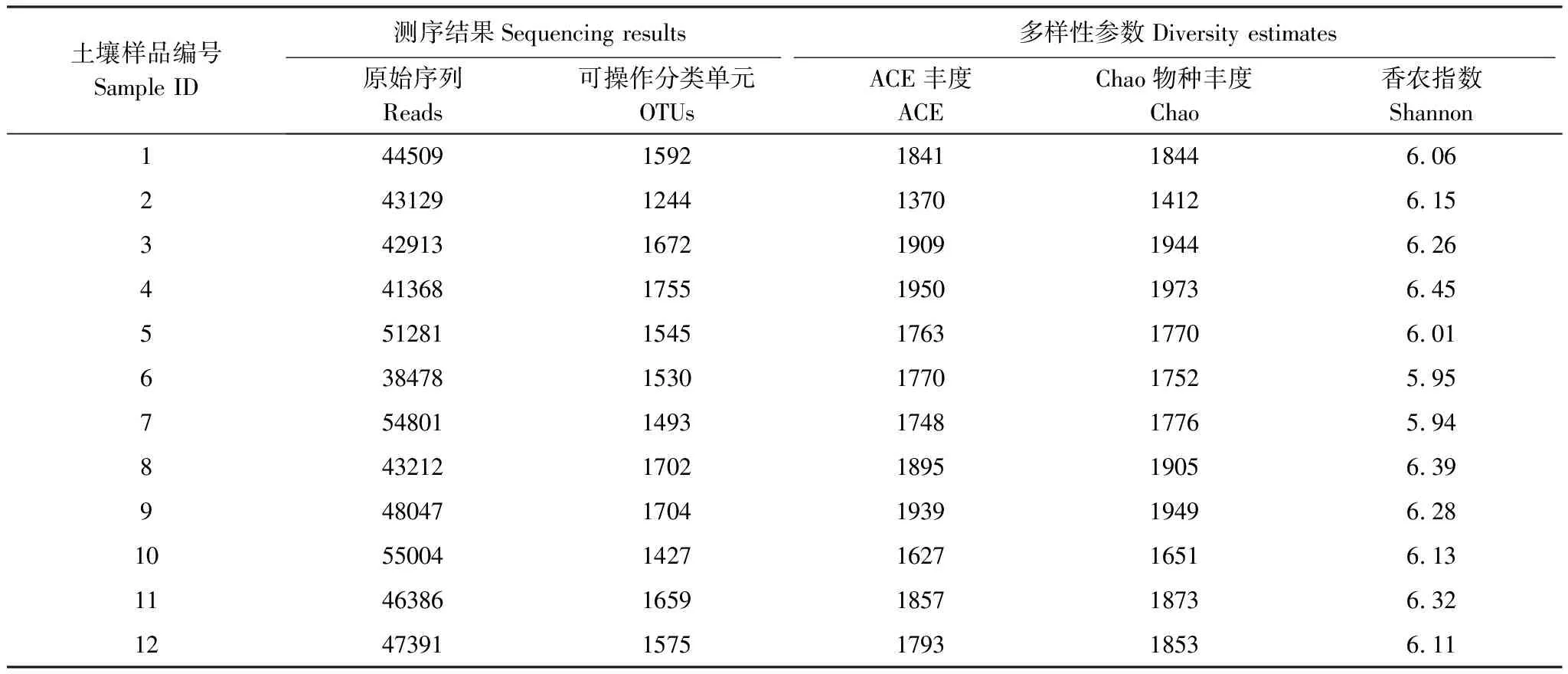
表2 土壤样本Miseq测序结果以及采样区域的细菌多样性评估
编号1—3:土样来自基向处理组根状茎连接的遮荫近端分株;编号4—6:土样来自基向处理组根状茎断开的遮荫近端分株;编号7—9:土样来自顶向处理组根状茎连接的遮荫远端分株;编号10—12:土样来自顶向处理组根状茎割断的遮荫远端分株

图4 所有样本基于OTU数目的稀释性曲线(97%相似度) Fig.4 Rarefaction curves of the OTU number at 97% similarity for every soil sample
序列比对结果表明,所有土壤样本的OTUs归属于13个门(phylum),419个属(genus)(相对丰度低于1%的菌群归于others组中)(图5,图6,表2)。其中变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)在所有样品均占据较大比重。热孢菌门(Thermotogae)细菌仅在顶向处理组的白夹竹遮荫分株根际土壤中被发现,在基向处理组则未发现。
Xanthobacteraceae(uncultured)包含一属Azorhizobium,具有共生固氮能力,其在顶向处理组中最大丰度为2.05%,在基向处理组中最大丰度为2.99%(图5,图6)。另一种固氮菌属Bradyrhizobium,相对丰度最高为3.07%。Nitrosomonadaceae(uncultured)隶属β-变形菌纲,该科包含的亚硝化单胞菌属(Nitrosomonas)以及亚硝化螺菌属(Nitrosospira)土壤细菌调控着铵盐向亚硝酸盐转化的氨氧化过程,是土壤生态环境中重要的氨氧化细菌(AOB)[23]。本研究中,顶向处理组中其相对丰度从1.17% 到 3.96%,而基向处理组中其相对丰度较高(2.43%—11.80%)(图5,图6)。
在所有的土壤样品均检测出了硝化螺菌门(Nitrospirae)的土壤微生物,相对丰度1.77%—7.83%不等(图5,表3)。该门仅包含一单科硝化螺菌科(Nitrospiraceae),该科的硝化螺菌属(Nitrospira)可将亚硝酸盐转化成硝酸盐,是N素循环过程中重要的硝化细菌[24]。顶向处理组中,Nitrospira相对丰度从0.77%到2.71%,基向处理组中其相对丰度从1.76%到5.63%不等(图5,图6)。Nitrospinaceae(uncultured)在土壤样品2中发现(相对丰度1.41%)(图6)。Nitrospira和Nitrospinaceae均是重要的亚硝酸盐氧化细菌(NOB)[25]。
3 讨论
光合产物等物质在克隆分株间的运输(顶向、基向传输)对克隆植物生长有着重要生态意义,特别是处于资源异质性斑块下的克隆植物[26]。已有研究表明,生长在适宜生境斑块的克隆分株会传输新形成的同化产物给生境条件相对恶劣斑块中的分株,以缓解其受到限制的光合能力或养分摄取能力[27]。本研究中,根状茎连接的遮荫分株根际土壤较高的DOC含量,表明克隆整合提高了白夹竹遮荫分株根际土壤的C有效性,暗示了白夹竹分株间克隆整合的存在。在树木环割实验中,由于新形成的光合产物向根系的传输被打断,从而削弱了根际沉积[28]。克隆整合对易分解碳的影响与环割实验的影响是相似的,即根状茎割断处理的遮荫分株(远端分株,近端分株)根际土壤的DOC以及MBC明显降低。同样地,先前的一项匍匐茎草本克隆植物的研究也取得了相似的结果[29]。
土壤微生物产生的胞外酶(如纤维素酶、木质素酶等)能调控土壤有机质降解、转移和矿化等过程[30]。NAGase参与的壳多糖水解[31],是土壤中有机碳的重要来源之一[32]。细菌和真菌均可以产生NAGase[16]。Urease主要由细菌、丝状真菌以及酵母菌产生,其在N素矿化过程中起着重要作用[33-34]。因此,相比割断分株,克隆整合导致的遮荫分株(远端分株,近端分株)根际土壤较高的微生物生物量(MBC、MBN)可能是上述两种水解酶活性增强的重要原因。同时,根状茎连接状态、同化产物传输的方向性对土壤胞外酶显著影响表明克隆整合的方向性对土壤有机质降解可能产生不同的影响(表1),个中机制则需更多研究证明。
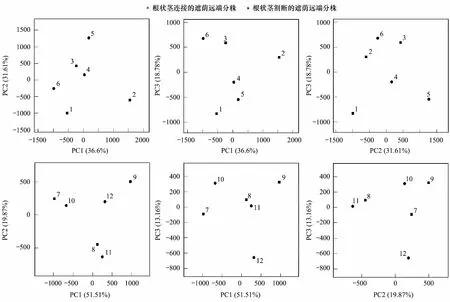
图7 基于属水平的土壤细菌群落组成的主成分分析图Fig.7 Microbial community composition described by principal component analysis (PCA) based on genus level information
主成分分析(PCA)作为一种对原有的复杂数据降维、简化、分析的技术,是分析环境微生物群落结构变化的重要手段[35]。基于OTU水平的PCA分析表明,无论是在顶向处理中还是在基向处理中,根状茎连接处理与割断处理下的遮荫分株根际土壤拥有相似的细菌群落结构。先前关于匍匐茎克隆植物Glechomalongituba生理整合的研究结果表明,克隆整合显著改变了遮荫分株根际土壤微生物群落组成,进而促进了根际生物过程[25,36]。而我们的研究结果与之不同。根际土壤C投入将会导致土壤有机质周转、微生物活性的短期变化[37],而土壤微生物群落种类多、数量大,土壤生态系统似乎存在一定程度的功能冗余(redundancy of function),如,微生物多样性与有机质降解之间并不存在直接的联系。某种土壤微生物数量的削减对特定的土壤生物过程造成的影响微乎其微,因为其他土壤微生物可能也执行这种功能[38]。这种功能冗余可能作为一种保障策略(insurance hypothesis),在不稳定的生态系统中,这些“冗余”物种可能起到维持系统生态平衡的作用[39]。因此,功能“冗余种”的存在可能使得土壤细菌群落保持较高的稳定性[40-41](resilience of soil microbial community),从而造成土壤细菌群落对克隆整合造成的根际土壤C投入响应“迟钝”。这是一种可能性的解释,而针对特定细菌功能群多样性的研究可能会提供进一步的实验证据。同时,相比传统指纹图谱(DGGE,PLFA)技术,单个样本的高测序通量及各分类水平上的高灵敏度使得高通量测序在环境微生物研究中发挥着越来越重要的作用[42]。虽然,高通量测序在广度、深度及分辨率上提高了多样性分析水平,并能够从整体群落层次上分析微生物遗传多样性[43]。但是,高通量测序得到的海量的序列数据可能不利于清晰阐释不同生态过程下的微生物群落结构的差异[11],这可能是本研究中克隆整合作用并没有对根际土壤细菌群落结构产生显著性影响的另一种解释。
克隆整合显著促进了白夹竹遮荫分株(远端分株,近端分株)的根际土壤C有效性,进而提高了根际微生物生物量。高通量测序能够较为全面、准确地反映根际细菌数量、活性变化影响的土壤细菌群落结构,并能够从整体细菌群落水平反映生物多样性。海量的序列数据可能不利于清晰阐释不同生态过程下的微生物群落结构差异。微生物多样性与土壤生态系统功能实现间关联研究的重点在于探讨多样性与微生物群落结构之间的关系以及群落结构与功能之间的关系[44]。因此,特定功能群(specific functional group)的定量化与高通量测序相结合,靶定丰度较低但发挥至关重要功能的微生物种群,可能为微生物生态学的研究提供新的思路。
致谢:感谢四川农业大学风景园林学院刘光立老师、小麦所彭远英老师在后期数据分析中提供的帮助。
参考文献(References):
[1] 董鸣. 克隆植物生态学. 北京:科学出版社, 2011:1- 20.
[2] Stuefer J F. Potential and Limitations of Current Concepts regarding the Response of Clonal Plants to Environmental Heterogeneity. Plant Ecology, 1996, 127(1):55- 70.
[3] Kuzyakov Y. Review: Factors affecting rhizosphere priming effects. Journal of Plant Nutrition and Soil Science, 2002, 165(4):382- 396.
[4] Rajaniemi T K, Allison V J. Abiotic conditions and plant cover differentially affect microbial biomass and community composition on dune gradients. Soil Biology and Biochemistry, 2009, 41(1):102- 109.
[5] Sinsabaugh R L, Antibus R K, Linkins A E. An enzymic approach to the analysis of microbial activity during plant litter decomposition. Agriculture Ecosystems and Environment, 1991, 34(1/4):43- 54.
[6] Sinsabaugh R L. Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biology and Biochemistry, 2010, 42(3):391- 404.
[7] Aon M A, Colaneri A C. II. Temporal and spatial evolution of enzymatic activities and physico-chemical properties in an agricultural soil. Applied Soil Ecology, 2001, 18(3):255- 270.
[8] 蒋婧, 宋明华. 植物与土壤微生物在调控生态系统养分循环中的作用.植物生态学报, 2010, 34(8):979- 988.
[9] Torsvik V, Sørheim R, Goksøyr J. Total bacterial diversity in soil and sediment communities—A review. Journal of Industrial Microbiology & Biotechnology, 1996, 17(3/4):170- 178.
[10] 周琳, 张杰. 群落分析中的16S rRNA及其基因16S rDNA优化扩增. 微生物学报, 2010, 50(1):7- 14.
[11] 夏围围, 贾仲君. 高通量测序和DGGE分析土壤微生物群落的技术评价. 微生物学报, 2014, 54(12):1489- 1499.
[12] 刘波, 胡桂萍, 郑雪芳, 张建福, 谢华安. 利用磷脂脂肪酸(PLFAs)生物标记法分析水稻根际土壤微生物多样性. 中国水稻科学, 2010, 24(3):278- 288.
[13] Fujii Y, Furubayashi A, Hiradate S. Rhizosphere soil method: a new bioassay to evaluate allelopathy in the field. //Proceedings of the 4th World Congress on Allelopathy, "Establishing the Scientific Base", New South Wales, Australia: Charles Sturt University, 2005:490- 492.
[14] Witt C, Gaunt J L, Galicia C C, Ottow J C G, Neue H U. A rapid chloroform-fumigation extraction method for measuring soil microbial biomass carbon and nitrogen in flooded rice soils. Biology and Fertility of Soils, 2000, 30(5/6):510- 519.
[15] Wu J, Joergensen R G, Pommerening B, Chaussod R, Brookes P C. Measurement of soil microbial biomass C by fumigation-extraction—an automated procedure. Soil Biology and Biochemistry, 1990, 22(8):1167- 1169.
[16] Parham J A, Deng S P. Detection, quantification and characterization of β-glucosaminidase activity in soil. Soil Biology and Biochemistry, 2000, 32(8/9):1183- 1190.
[17] Kandeler E, Gerber H. Short-term assay of soil urease activity using colorimetric determination of ammonium. Biology and Fertility of Soils, 1988, 6(1):68- 72.
[18] Dennis K L, Wang Y W, Blatner N R, Wang S Y, Saadalla A, Trudeau E, Roers A, Weaver C T, Lee J J, Gilbert J A, Chang E B, Khazaie K. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL- 10 producing T-cells. Cancer Research, 2013, 73(19):5905- 5913.
[19] Li Z P, Zhang Z G, Xu C, Zhao J B, Liu H L, Fan Z Y, Yang F H, Wright A D G, Li G Y. Bacteria and methanogens differ along the gastrointestinal tract of Chinese roe deer (Capreoluspygargus). PLoS One, 2014, 9(12):e114513.
[20] Fadrosh D W, Ma B, Gajer P, Sengamalay N, Ott S, Brotman R M, Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome, 2014, 2:6.
[21] Hong C, Si Y X, Xing Y, Li Y. Illumina MiSeq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas. Environmental Science and Pollution Research, 2015, 22(14):10788- 10799.
[22] Amato K R, Yeoman C J, Kent A, Righini N, Carbonero F, Estrada A, Gaskins H R, Stumpf R M, Yildirim S, Torralba M, Gillis M, Wilson B A, Nelson K E, White B A, Leigh S R. Habitat degradation impacts black howler monkey (Alouattapigra) gastrointestinal microbiomes. The ISME Journal, 2013, 7(7):1344- 1353.
[23] Bernhard A E, Donn T, Giblin A E, Stahl D A. Loss of diversity of ammonia-oxidizing bacteria correlates with increasing salinity in an estuary system. Environmental Microbiology, 2005, 7(9):1289- 1297.
[24] Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, Rattei T, Damsté J S S, Spieck E, Paslier D L, Daims H. ANitrospirametagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(30):13479- 13484.
[25] 闫媛, 黎力, 王亚宜, 林喜茂, 马骁, 陈玉. 采用高通量测序分析全程自养脱氮(CANON)系统不同脱氮效能下的微生物群落结构. 北京工业大学学报, 2015, 41(10):1485- 1492.
[26] Saitoh T, Seiwa K, Nishiwaki A. Importance of physiological integration of dwarf bamboo to persistence in forest understorey: a field experiment. Journal of Ecology, 2002, 90(1):78- 85.
[27] Saitoh T, Seiwa K, Nishiwaki A. Effects of Resource Heterogeneity on Nitrogen Translocation within Clonal Fragments ofSasapalmata: an Isotopic (15N) Assessment. Annals of Botany, 2006, 98(3):657- 663.
[28] Chen D M, Zhou L X, Wu J P, Hsu J, Lin Y B, Fu S L. Tree girdling affects the soil microbial community by modifying resource availability in two subtropical plantations. Applied Soil Ecology, 2012, 53(1):108- 115.
[29] Chen J S, Li J, Zhang Y, Zong H, Lei N F. Clonal integration ameliorates the carbon accumulation capacity of a stoloniferous herb,Glechomalongituba, growing in heterogenous light conditions by facilitating nitrogen assimilation in the rhizosphere. Annals of Botany, 2015, 115(1):127- 136.
[30] Schimel J P, Weintraub M N. The implications of exoenzyme activity on microbial carbon and nitrogen limitation in soil: a theoretical model. Soil Biology and Biochemistry, 2003, 35(4):549- 563.
[31] Sinsabaugh R L, Antibus R K, Linkins A E, McClaugherty C A, Rayburn L, Repert D, Weiland T. Wood Decomposition: Nitrogen and Phosphorus Dynamics in Relation to Extracellular Enzyme Activity. Ecology, 1993, 74(5):1586- 1593.
[32] Smucker R A, Kim C K. Chitinase induction in an estuarine system// Llewellyn GC, O′Rear CE, eds. Biodeterioration research. New York: Plenum Press, 1987:347- 355.
[33] Gianfreda L, Rao M A, Piotrowska A, Palumbo G, Colombo C. Soil enzyme activities as affected by anthropogenic alterations: intensive agricultural practices and organic pollution. Science of the Total Environment, 2005, 341(1/3):265- 279.
[34] Mobley H L, Hausinger R P. Microbial ureases: significance, regulation, and molecular characterization. Microbiological Reviews, 1989, 53(1):85- 108.
[35] Wang Y, Sheng H F, He Y, Wu J Y, Jiang Y X, Tam N F Y, Zhou H W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags. Applied and Environmental Microbiology, 2012, 78(23):8264- 8271.
[36] Lei N F, Li J, Ni S J, Chen J S. Effects of Clonal Integration on Microbial Community Composition and Processes in the Rhizosphere of the Stoloniferous HerbGlechomalongituba(Nakai) Kuprian. PLoS One, 2014, 9(9):e108259.
[37] Kuzyakov Y, Friedel J K, Stahr K. Review of mechanisms and quantification of priming effects. Soil Biology and Biochemistry, 2000, 32(11/12):1485-1498.
[38] Nannipieri P, Ascher J, Ceccherini M T, Landi L, Pietramellara G, Renella G. Microbial diversity and soil functions. European Journal of Soil Science, 2003, 54(4):655- 670.
[39] Loreau M, Naeem S, Inchausti P, Bengtsson J, Grime J P, Hector A, Hooper D U, Huston M A, Raffaelli D, Schmid B, Tilman D, Wardle D A. Biodiversity and ecosystem functioning: current knowledge and future challenges. Science, 2001, 294(5543): 804- 808.
[40] Griffiths B S, Philippot L. Insights into the resistance and resilience of the soil microbial community. Fems Microbiology Reviews, 2013, 37(2):112- 129.
[41] Cruzmartínez K, Suttle K B, Brodie E L, Power M E, Andersen G L, Banfield J F. Despite strong seasonal responses, soil microbial consortia are more resilient to long-term changes in rainfall than overlying grassland. The ISME Journal, 2009, 3(6):738- 744.
[42] 楼骏, 柳勇, 李延. 高通量测序技术在土壤微生物多样性研究中的研究进展. 中国农学通报, 2014, 30(15):256- 260.
[43] 姬洪飞, 王颖. 分子生物学方法在环境微生物生态学中的应用研究进展. 生态学报, 2016, 36(24):8234- 8243.
[44] O′Donnell A G, Seasman M, Macrae A, Waite L, Davies J T. Plants and fertilisers as drivers of change in microbial community structure and function in soils. Plant and Soil, 2001, 232(1/2): 135- 145.
