超高效液相色谱-串联质谱法测定动物源性食品中12种青霉素类药物残留量
2018-06-14梁晶晶沈丹徐潇颖丁宇琦罗金文
梁晶晶,沈丹,徐潇颖,丁宇琦,罗金文*
1(浙江省食品药品检验研究院,浙江 杭州, 310052) 2(通标标准技术服务有限公司,浙江 杭州, 310052)
青霉素族属β-内酰胺类抗生素,通过抑制细菌细胞壁四肽侧链和五肽交联桥的结合来阻碍细胞壁合成,具有较强的杀菌作用,广泛应用于人和动物尿道、胃肠道和呼吸道感染等疾病[1-2]。然而,部分养殖户因其价格低廉、使用方便,在养殖过程中滥用青霉素类药物,导致其在动物组织中蓄积残留。青霉素在动物源性食品中的残留会危害人体健康,破坏人类肠道的正常菌群环境,导致人体免疫力下降。为确保消费者的安全,我国及欧盟等组织均对动物组织中的青霉素类药物残留限量做了严格的规定。如我国规定所有动物的肌肉、脂肪、肝、肾食品中阿莫西林和氨苄西林的最高残留限量为50 μg/kg,氯唑西林和苯唑西林的最高残留限量300 μg/kg[3]。青霉素药物残留依然是人们关注的热点。
目前,检测青霉素类抗生素的方法主要有酶联免疫法[4-5]、液相色谱法[6-8]、液相色谱-串联质谱法等[9-11]。酶联免疫法属于半定量筛查方法,容易产生假阳性现象;液相色谱法检测抗生素, 定性效果差,且不适合多残留同步分析。近年来,液相色谱-串联四极杆质谱联用(liquid chromatography-tandem mass spectrometry, LC-MS/MS)技术由于兼具液相色谱的分离能力和串联质谱的高灵敏度、高专属性特点,正逐渐成为兽药残留分析的有效手段,但现有的方法都存在前处理步骤繁琐,测定周期长等缺点。
PRiME HLB是一种最新型的固相萃取柱,是基于“N-乙烯吡咯烷酮二乙烯基苯聚合物”填料的反相复合填料,该复合填料经过特殊设计,对于动物源性食品中的蛋白质、脂肪和磷脂等物质具有特异性的吸附能力,且无需活化与平衡,样品经有机溶剂提取后,可直接上样到小柱上,操作极其简便,不会出现堵柱情况,最多只需3个步骤即可完成样品制备过程,净化速度可提高40%,净化后,可以去除动物源性食品中蛋白、脂肪和磷脂等95%以上的基质干扰物,从而确保数据的稳定性和可靠性。此方法只需采用最简单的过滤式样品制备流程即可实现最洁净的样品净化目标,操作简单、省时快速。本研究采用PRiME HLB样品净化技术,超高效液相色谱-串联四级杆质谱(UPLC-MS/MS)分析手段,测定食品中12种青霉素族抗生素类药物残留量的检测方法。
1 材料与方法
1.1 仪器及材料
AB5500液相色谱-串联质谱仪(美国 SCIEX);甲醇,乙腈,甲酸(均为色谱纯);PRiME HLB固相萃取柱(Waters公司);羟氨苄青霉素、氨苄青霉素、苄青霉素、苯氧甲基青霉素、苯唑青霉素、邻氯青霉素、乙氧萘青霉素和双氯青霉(Dr.Ehrenstorfer);甲氧苯青霉素和哌拉西林(USP),苯咪青霉素(北京振翔),氘代青霉素G(WITEGE)。
实验样品:鸡和鲈鱼,取鸡胸肉和鲈鱼的肌肉部位进行试验。
1.2 实验方法
1.2.1 标准溶液配制
对照品储备液配制:分别精密称取适量的标准品(羟氨苄青霉素、氨苄青霉素、苯咪青霉素、甲氧苯青霉素、苄青霉素、苯氧甲基青霉素、苯唑青霉素、苯氧乙基青霉素、邻氯青霉素、乙氧萘青霉素、双氯青霉素、哌拉西林),分别用30%乙腈水溶解并定容至25 mL,各青霉素类抗生素储备液浓度为500 μg/mL,于-18 ℃下存放。
混合标准中间溶液的配制:分别精密移取适量对照品储备液,用水配制成约500 ng/mL的混合标准中间溶液。
同位素内标储备液:取适量的氘代青霉素G盐,用30%乙腈水溶解并定容至25 mL,同位素内标储备液浓度为200 μg/mL,于-18 ℃下保存。
同位素内标工作液:用纯水将同位素内标储备液稀释500倍,配制成400 ng/mL的内标工作液。
1.2.2 样品前处理
精密称取2.5 g样品(准确至0.01 g)于50 mL离心管中,精密加入同位素内标工作液0.25 mL,加80%乙腈水溶液10 mL,涡旋混匀1 min,振荡器振荡30 min后,20 000 r/min离心10 min,上清液转移至20 mL容量瓶中,残渣用7 mL 80%乙腈水溶液重复提取1次,合并上清液,用80%乙腈水溶液定容至20 mL,取5.0 mL加载到6CC规格的PRiME HLB 固相萃取柱,保持1 s 1滴的流速,取4 mL流出液于37 ℃下氮吹至小于0.7 mL,残留液用纯水定容至1.00 mL,过0.22 mL微孔滤膜,上LC-MS/MS测试。
1.2.3 空白基质溶液的制备
取不含青霉素类药物的空白样品,除不加内标工作液外,同1.2.2处理,制得空白样品基质溶液。
1.2.4 标准曲线的制备
精密移取标准中间溶液和内标工作液适量(标准工作溶液中内标物浓度应与样品待测液中一致),用空白样品基质溶液配制成5~100 ng/mL的系列混合标准工作溶液。
1.2.5 色谱与质谱条件
液相采用CORTECS C18色谱柱(100 mm×2.1 mm,2.7 μm);流动相为A:0.1%甲酸水,B:甲醇;梯度洗脱程序(B变化):0~3 min,15%~50%;3~6 min,50%~70%;6~8 min,保持70%,8~8.1 min,70%~15%,8.1~12 min,保持15%;流速:0.4 mL/min;柱温:35 ℃;进样量:10 μL。
质谱采用电喷雾离子源,正离子模式;电离电压:5 500 v;离子源温度:500 ℃;气帘气流速:30 L/min;碰撞气流量:30 L/min。

表1 质谱参数Table 1 MS/MS parameters
2 结果与讨论
2.1 色谱条件的选择
CORTECS C18色谱柱采用不同键合相颗粒特性以及表面带电修饰技术,具有柱效高、选择性强,峰形好等优点,本实验以CORTECS C18(100 mm×2.1 mm,2.7 μm)作为分析柱,对比了甲醇和乙腈作为有机相时的分离效果,发现用甲醇作为有机相时各目标化合物的分离效果更好,因此选择甲醇作为有机相。ESI电离是在溶液状态电离,而在水相中加入0.1%甲酸时,各化合物在ESI+模式下的离子化效果更好,峰形有所改善,灵敏度也相应提高,因此本实验选取甲醇及0.1%甲酸水作为流动相。经甲醇-水梯度洗脱所得的多反应监测离子流色谱图见图1。

图1 标准品多反应监测离子流色谱图Fig.1 Multiple reaction monitoring (MRM) chromatograms of mixed standards solution
2.2 提取溶剂和复溶溶液的选择
因考虑到甲醇的化学结构中含有羟基,会加速青霉素降解为青霉噻唑酸酯[12],故选择乙腈作为提取溶剂,预实验采用70%乙腈水和70%乙腈水(含0.1%甲酸)2种溶液作为提取剂,结果表明,用70%乙腈水(含0.1%甲酸)提取后,部分化合物回收率较低,不到60%,如阿莫西林、苯氧乙基青霉素,分析原因,由于多种青霉素类药物在酸性条件下不稳定所致。随后考察了70%乙腈水、80%乙腈水和90%乙腈水3种不同配比的提取剂对提取率的影响,结果表明,80%乙腈水对于各个化合物的提取效率最好,都能达到80%以上,沉淀蛋白完全,易于离心分离。因此,提取溶剂采用80%乙腈水,不加酸。
复溶溶液的选择:对比了30%乙腈水溶液、30%甲醇水溶液和纯水作为复溶液的色谱图,发现采用30%乙腈水和30%甲醇水溶液复溶后,阿莫西林产生较强的溶剂效应,呈现双峰,而用纯水复溶后的阿莫西林峰形较好,因此,选择纯水作为复溶液。
2.3 氮气浓缩条件的选择
为了对净化后的提取液进行浓缩,提高检测灵敏度,需对提取液进行氮吹。由于青霉素类药物具有光不稳定性和热不稳定性的特点,在前处理操作中应尽量避光、避热,并且在较短的时间内完成前处理过程,时间过长,温度过高都会影响稳定性,加速其降解,因此,需对氮吹的温度进行研究与优化。本实验考察了30、37和45 ℃ 3个氮吹温度条件下各个化合物的稳定性,结果表明,在30 ℃和45 ℃条件下,部分化合物有不同程度的降解,导致回收率在60%~80%,而在37 ℃条件下则可达到80%以上的回收率,认为是由于30℃温度虽低,但导致氮吹时间过长,以及45 ℃温度过高加速降解所致。因此,本实验选择37 ℃作为氮吹的最佳温度。
2.4 方法学评价
为了消除基质效应对测定结果的影响,本方法采用基质标准曲线对青霉素类药物进行分析。按照优化后的色谱条件对12种青霉素类药物做基质标准曲线,并按国家标准和行业标准规定的检出限含量(见表2)做基质加标。结果表明12种青霉素在各自浓度范围内线性关系良好,相关系数r2≥0.998,信噪比均大于3,说明该方法检出限能达到国标方法规定的检出限。
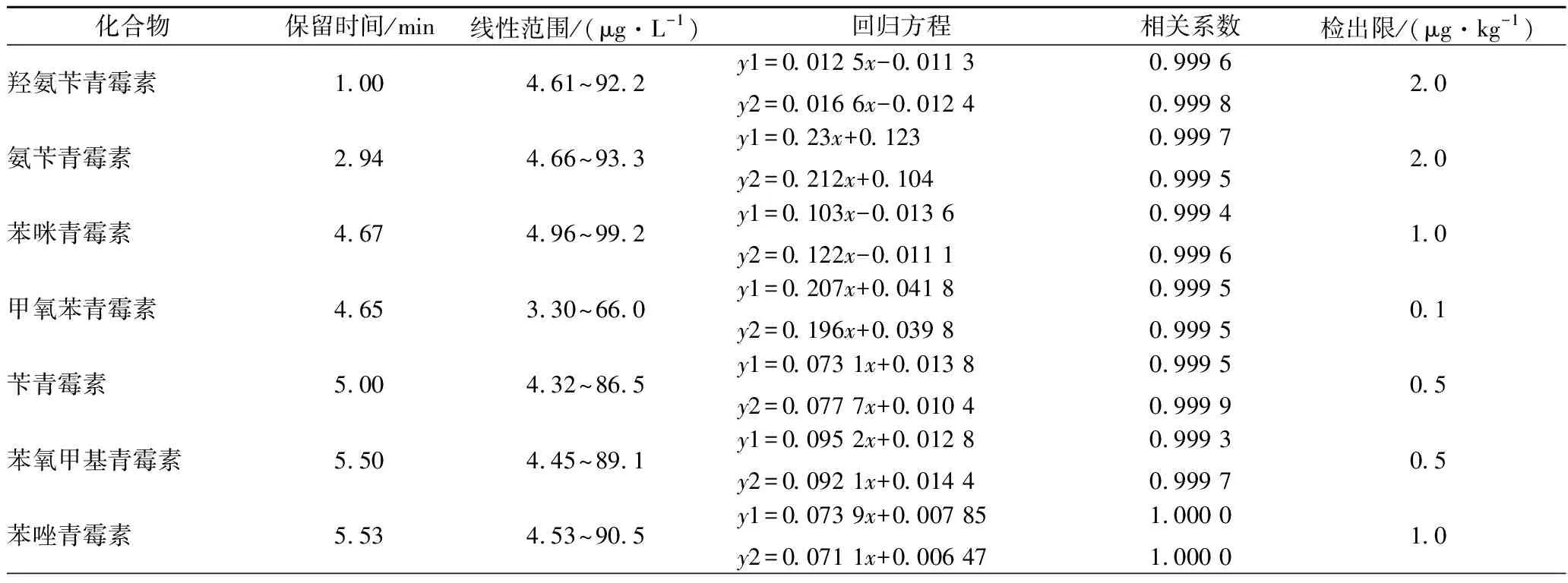
表2 12种青霉素族抗生素测定的线性方程、相关系数和检出限Table 2 Linear equations, correlation coefficient and limit of detection of 12 penicillins antibiotics
续表2

化合物保留时间/min线性范围/(μg·L-1)回归方程相关系数检出限/(μg·kg-1)苯氧乙基青霉素5.817.26~145.2y1=0.038 2x-0.018 9y2=0.041 1x-0.014 40.999 80.999 910.0邻氯青霉素5.724.22~84.4y1=0.053 4x-0.035 7y2=0.056 9x-0.042 60.999 30.999 41.0乙氧萘青霉素6.153.91~78.2y1=0.26x-0.128y2=0.271x-0.1550.999 80.999 70.25双氯青霉素6.094.88~97.6y1=0.017 6x-0.006 21y2=0.015 7x-0.005 990.999 70.999 92.0哌拉西林4.825.27~105.4y1=0.061 2x-0.013 6y2=0.059 8x-0.016 90.998 90.999 31.0
注:y1代表以鸡肉为基质的回归方程,y2代表以鱼肉为基质的回归方程。
分别对鸡肉和鱼肉(均为阴性样品)进行不同水平的加标回收试验,加标浓度分别为20、50、150 μg/kg,每一浓度进行6组平行实验,计算回收率及相对标准偏差,结果见表3。由表3可知,各组分的平均回收率在81.7%~111.9%,相对标准偏差在0.89%~8.56%,说明该方法具有良好的准确性和精密度。
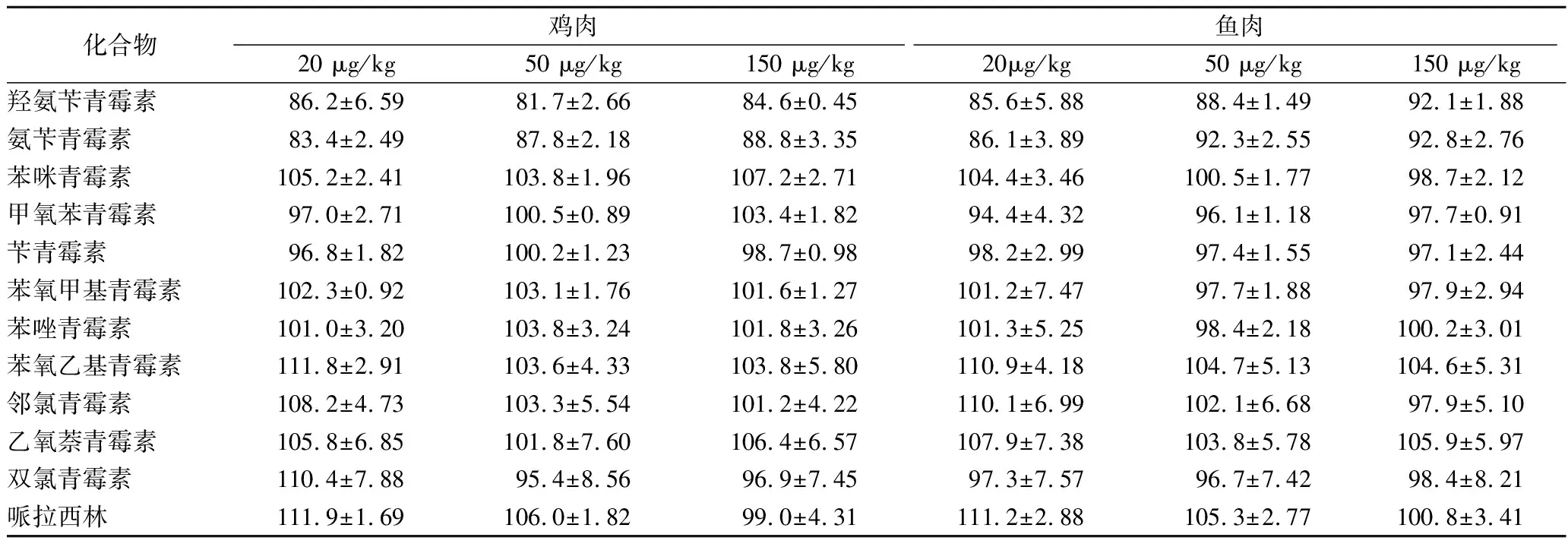
表3 12种青霉素族抗生素测定的回收率和精密度Table 3 recovery and precision of 12 penicillins antibiotics
2.5 实际样品测定
从市场上抽取鸡肉、猪肉和鱼各10份,利用本方法对其进行青霉素类药物残留检测,测定结果均为阴性。
3 结论
本实验应用PRiME HLB净化技术,采用超高效液相色谱-四级杆串联质谱联用方法建立了动物源性食品中青霉素族抗生素类药物残留的检测方法,各组分在各自浓度范围内线性关系良好,相关系数r2≥0.998,平均回收率在81.7%~111.9%,相对标准偏差在0.89%~8.56%,方法检出限达到标准规定的检出限含量。因此,本方法用于动物源性食品中青霉素族抗生素药物残留检测,具有准确、快速、简便、灵敏度高等优点。
[1] 陈卓婧,阎淑男,夏澜,等.农产品中青霉素残留检测技术研究进展[J].浙江农业科学,2013,11:1 476-1 479.
[2] HOU J P,POOLE J W.Lactam antibiotics:their physicochemical properties and biological activities in relation to structure[J].J.Pharm.Sci.,1971,60(4):503-532.
[3] 动物性食品中兽药最高残留限量.农业部235号公告,2002.
[4] 姜侃,厉永纲,沈泓,等.应用酶联免疫技术建立鲜乳中β-内酰胺酶间接检测方法[J].中国食品卫生杂志, 2013 , 25(5):405-409.
[5] SAMSONOVA,ZHVH,SHCHELOKOVA O S,IVANOVA N L,et aL.Enzyme immunoassay of ampicillin in milk [J].Pfikl Biokhim Mikrobiol,2005,41(6):668.
[6] 牛奶中青霉素类药物残留量的检测方法-高效液相色谱法.农业部781号公告,2006.
[7] GB 29682—2013.食品安全国家标准 水产品中青霉素类药物多残留的测定 高效液相色谱法[S].
[8] ANG C Y,LUO W.Rapid determination of ampicillin in bovine milk by liquid chromatography with fluorescence detection [J].JA O A CInt,1997,80(1):25-30.
[9] GB/T 22952—2008.河豚鱼和鳗鱼中阿莫西林、氨苄西林、哌拉西林、青霉素G、青霉素V、苯唑西林、氯唑西林、萘夫西林、双氯西林残留量的测定 液相色谱-串联质谱法[S].
[10] GB/T 18932.25—2005.蜂蜜中青霉素G、青霉素V、乙氧萘青霉素、苯唑青霉素、邻氯青霉素、双氯青霉素残留量的测定方法 液相色谱-串联质谱法[S].
[11] 郭萌萌,李兆新,谭志军,等.分散固相萃取/液相色谱-串联质谱法测定水产品中的8种青霉素残留[J].分析测试学报, 2011, 30(9):969-975.
[12] 冯月超 ,范筱京,贾丽,等.液相色谱一质谱联用法测定牛奶中14种青霉素及相应青霉噻唑酸残留量[J].分析试验室, 2012(9): 67-70.
