基于CRISPR/Cas9系统的HEK293T细胞DMD基因第51号外显子的靶向敲除
2018-06-14马珊珊崔思颖屈素真蔡奥捷关方霞孔祥东
李 双,马珊珊,崔思颖,屈素真,蔡奥捷,关方霞*,孔祥东*
(1.郑州大学第一附属医院 遗传与产前诊断中心, 河南 郑州 450052; 2.郑州大学 生命科学学院, 河南 郑州 450001)
杜氏肌营养不良(Duchenne muscular dystrophy,DMD,OMIM#310200)是最常见的神经肌肉系统遗传病,为X-连锁隐性,由DMD基因突变引起抗肌萎缩蛋白(dystrophin)合成障碍, 造成肌肉收缩不良[1], 患
者常于20岁左右死于呼衰或心衰,临床尚无有效治疗方法。
DMD基因含79个外显子,统计显示约80%的DMD患者可通过跳跃特定外显子修复阅读框,恢复部分dystrophin表达[2],其中可跳跃51外显子(exon51)者约13%[3]。基因编辑可从根本上修复突变基因,并通过细胞分裂稳定传递。成簇规律间隔短回文重复序列系统(clustered regularly interspaced short palindromic repeats, associated RNA guided endonuclease Cas9, CRISPR/Cas9) 通过sgRNA(singe-guide RNA)5′端识别DNA,可实现对任意DNA序列的靶向修饰[4- 5]。若同时转入多个sgRNA,则可完成对多个靶向位点或大片段基因编辑[6- 7]。本研究使用人胚肾上皮(HEK293T)细胞,转入靶向DMD基因exon51两端的CRISPR/Cas9单载体质粒系统,实现对指定外显子的敲除并提高了敲除效率,为DMD的基因治疗及机制研究奠定了实验基础。
1 材料与方法
1.1 材料
HEK293T细胞为实验室所有;PX458/PX459质粒(Addgene质粒库);限制性内切酶BbsⅠ、XbaⅠ和磷酸化酶FastAP (Fermentas公司);连接酶Quick ligase(NEB公司);细胞转染试剂盒ViaFectTMTransfection Reagent (Promega公司);无内毒素质粒小提试剂盒和细胞基因组提取试剂盒(北京天根生物有限公司);DMEM高糖培养基和磷酸盐缓冲液(Thermo公司);胎牛血清(Gemini公司);T载体(TaKaRa公司);Detecase内切酶(上海吉凯基因公司);高保真KOD FX DNA扩增酶(Toyobo公司)。
1.2 方法
1.2.1 靶位点选择及引物设计:运用多重连接探针扩增技术(multiplex ligation-MLPA)对HEK293T细胞DMD基因进行检测,确认是否完整。
选取DMD基因exon51两端内含子区域序列,应用网站http://crispr. mit.edu/设计靶向sgRNA,每端选取3条分数较高者用于质粒构建。靶点设计遵循以下原则: 1)G必须出现在sgRNA模版DNA序列5′ 端,若不是则合成时另添加1个G;2)尽量避开GC富集区域;3)选择特异性位点。在正义链与反义链模板5′ 端分别添加CACC与AAAC,以分别与BbsⅠ酶切后形成的黏性末端互补。
应用Genetool设计引物,上下游引物分别距离靶序列两侧翼100~150 bp,扩增片段大小250~350 bp。将磷酸化的sgRNA双链及上述引物交由上海生工生物有限公司合成。
1.2.2 PX459- sgRNA重组质粒的构建:质粒载体系统(PX458/PX459)为化酿脓链球菌(S.Pyogenes)的CRISPR系统改造,靶向序列为20个碱基,下游PAM序列为NGG。其中PX458质粒带EGFP标签,PX459质粒带嘌呤霉素抗性基因。
37 ℃下使用核酸内切酶BbsⅠ线性化PX459,蒸馏水重悬磷酸化的sgRNA双链到终浓度并以1∶100稀释,使用Quick ligase试剂以sgRNA:线性载体DNA为3~10∶1比例4 ℃连接过夜,产物转化至Stbl3感受态细胞后摇菌,部分菌液送测序,余置于-80 ℃保存。
1.2.3 细胞的培养和转染:使用DMEM高糖培养基,10%胎牛血清,置于37 ℃、5% CO2细胞培养箱中培养HEK293T细胞。转染前24 h,以5×105个细胞/孔均匀铺至6孔板中,使转染时达50%~60%汇合;使用ViaFectTMTransfection Reagent在6孔板中分别转染5个重组质粒,等量PX458为阴性对照,24 h后加入嘌呤霉素(puro,puromycin)至浓度1 g/L,3 d后维持低浓度puro(0.5 g/L)继续培养2~3 d后提取细胞基因组DNA。
1.2.4 Surveyor法检测sgRNA切割效率:在PCR管中配置以下体系:DNA 2 μL,上下游引物各1 μL,2 ×PCR Mix 13 μL,ddH2O 8 μL。PCR程序:98 ℃ 3 min,30循环(98 ℃ 10 s,60 ℃ 15 s,72 ℃ 1 min),72 ℃ 10 min,98 ℃ 3 min。PCR扩增后,自然冷却至40 ℃以下,使得细胞内的突变片段互相杂交而形成错配。在灭菌PCR管中配置以下体系:PCR产物2~3 μL,Detecase缓冲液2 μL,Detecase 1 μL,ddH2O 至10 μL。45 ℃反应20 min后,立即在上述体系中加入2 μL stop缓冲液,进行聚丙烯酰氨(PAGE)凝胶电泳并银染,观察条带切割情况。
1.2.5 PX459- 双sgRNA重组质粒的构建:PCR扩增PX459- sgRNA5- 1质粒中U6启动子及sgRNA5- 1序列,使用核酸内切酶XbaⅠ处理后与同样XbaⅠ线性化的PX459- sgRNA3- 1质粒连接。将连接产物转化入感受态Stbl3中,涂抹于含氨苄的LB平板上,挑单克隆,摇菌,小提质粒后送测序验证,得到重组质粒PX459- 2sgRNA5- 1+3- 1。
1.2.6 双末端高效sgRNA共转染切割外显子:以细胞计数约为1×106个/孔将细胞均匀铺至3个60 mm培养皿中,待细胞达50%~60%汇合,A、B、C皿分别转染质粒PX459- 2sgRNA5- 1+3- 1,PX459- 2sgRNA5- 1,PX458(阴性对照)。转染24 h后,加入嘌呤霉素至浓度1 g/L,3 d后维持0.5 g/L puro继续培养2代后保留部分细胞,余提取基因组DNA。
使用高保真KOD酶对3组细胞DMD基因exon51进行扩增,扩增产物连接T载体并送测序。
2 结果
2.1 HEK293T细胞DMD基因检测及靶位点设计
MLPA结果显示DMD基因完整,无外显子缺失或重复(图1A);在DMD基因exon51两端靶点设计原理及具体序列(图1B)。
2.2 PX459质粒信息及插入片段
将添加了黏性末端的靶向exon51两端sgRNA分别命名为E51- 5- 1~3,E51- 3- 1~3(表1),验证质粒构建的通用引物hU6-primer及测序引物序列(表2)。
2.3 PX459- sgRNA质粒测结果及Surveyor分析
成功构建含靶向DMD基因exon51 5′端sgRNA的重组质粒2个,3′端3个(图2)。Surveyor法显示5个sgRNA均有切割活性,其中DMD- 5- 1和DMD- 3- 1活性最高(图3)。
分组实验显示DNA 2μg,Viafection 8μL时转染效率可高达80%,且使用转染试剂较少,可用于后续实验(图4)。
2.4 PCR及T载体测序分析
对6株连接成功的菌落进行测序,3株显示DMD基因exon51敲除,敲除效率达50%;其中2株在连接处发生随机插入,1株出现单个碱基缺失(图5)。
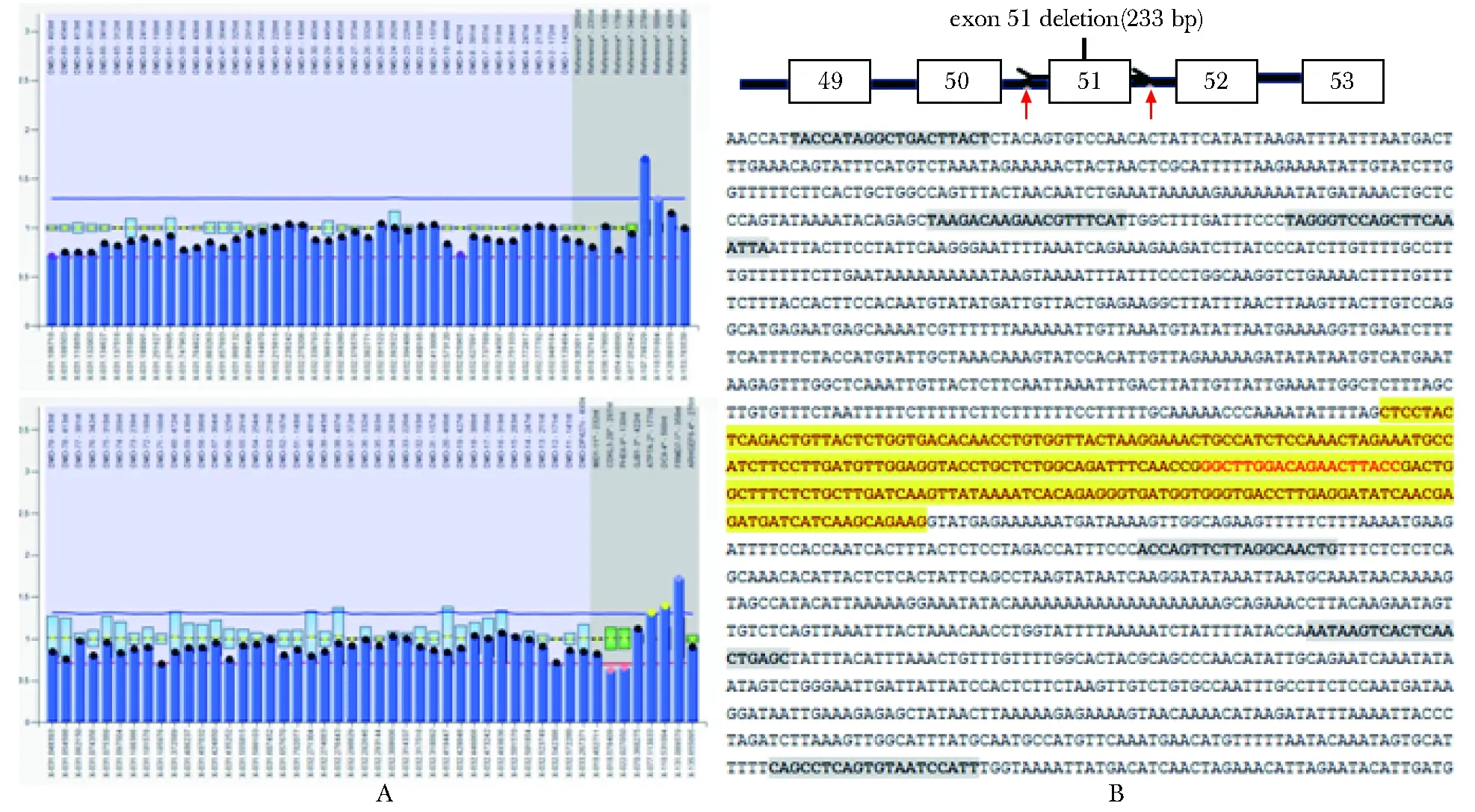
A.the MLPA result of DMD gene of HEK293T cells, normal columns reached around the blue line and exceeded the red line; B.diagram of exon51 knock-out, the arrows pointed the target cleavage sites cut by Cas9 protein under the guidance of double sgRNAs; the introns around the cleavage sites connected through non-homologous end joining; the sequence of exon51 was highlighted and the sgRNAs sequence were bolded

图1 MLPA检测及靶位点设计Fig 1 MLPA testing of DMD gene and target sites design
cohesive ends were represented by small letters.
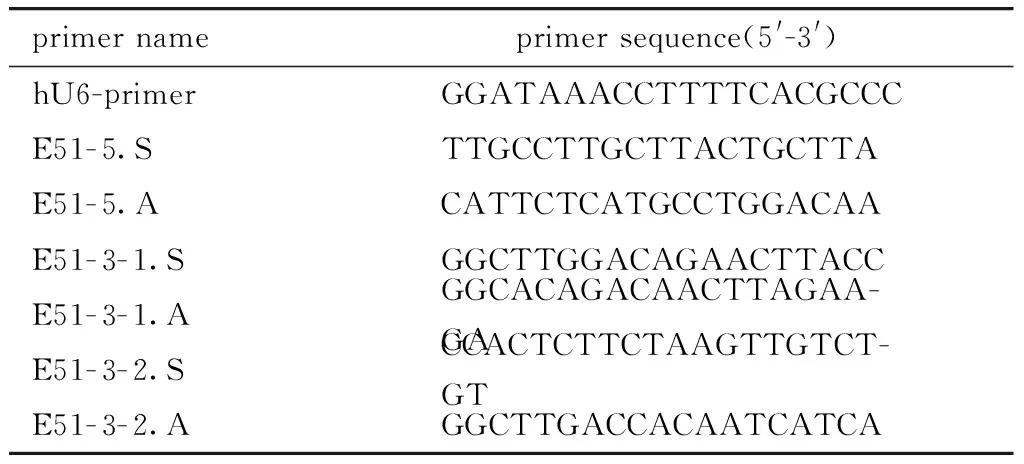
表2 验证质粒构建及sgRNA活性的引物序列

The segments cut by Surveyor endonuclease were pointed by arrows, indicting DNA mismatch; the high-efficiency sgRNAs used in further experiments were circled图3 sgRNA活性检测Fig 3 Activity detection of sgRNAs
3 讨论
DMD的基因治疗研究中,以慢病毒为载体导入minidystrophin研究因机体免疫反应在人体实验中失败[8];反义寡核苷酸介导的外显子跳跃治疗,长期疗效也有待验证[9]。基因编辑是修复基因突变的最根本方法,可实现突变修复的稳定传递,CRISPR/Cas9系统因简单、高效、安全性高和毒性小等特点,一经发现便被广泛采用。在DMD基因编辑治疗中,通过同源重组修复(homology-directed repair, HDR)插入缺失的外显子或修复点突变可完成精准的基因修复,但通常情况下HDR只存在于分裂细胞中;通过非同源重组修复(non-homologous endjoining, NHEJ)引入小的插入或缺失可修复读码框[10],但效果并不明确;而BMD患者体内可检测出缩短dystrophin 蛋白突变体[2],这本身已证明敲除特定外显子用以治疗DMD的可行性。突变热点(如exon45- 55,exon51)的存在,使得设计1次sgRNA可用以多个患者,减少了临床应用成本。2015年已有研究使用CRISPR/Cas9系统对DMD患者成肌细胞进行基因修饰,随后注射至DMD模型小鼠骨骼肌内,检测到正常的人dystrophin表达[11]。但如何提高切割及转染效率,降低脱靶,寻找合适的细胞来源依然是该领域的研究热点。
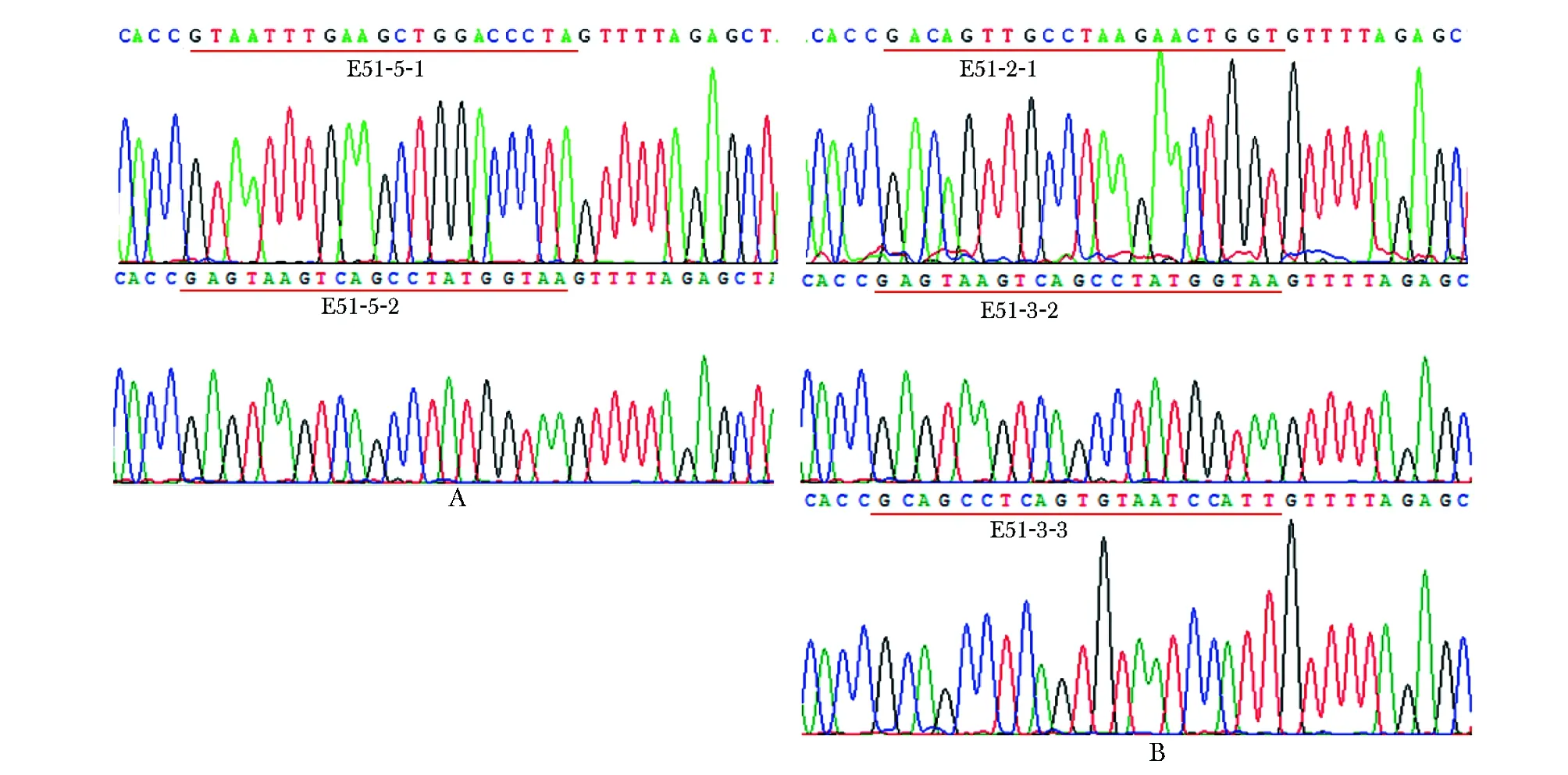
A.two plamsids are located on 5′ terminal; B.three plasmids are located on 3′ terminal; red lines marked the insertion sequence of sgRNA
图2PX459-sgRNA质粒测序图
Fig2SequencediagramofPX459-sgRNAplasmids

A.plasmids were transferred into cells by lipo2000; B, C, D.plasmids were transferred into cells by ViaFection; the top images were observed in white light and the bottom images were observed under fluorescence microscope
图46孔板中质粒转染HEK293T效果图
Fig4Comparisonoftransfectionefficiencyamongdifferentconditions

A.control grope, the cleavage site of E51- 5- 1 is complete; B, C.two base(GG/GC) are random inserted between the two cleavage sites; D.one base A is random missing between the two cleavage sites
图5T载体测序结果
Fig5ResultsofTvectorsequencing
多sgRNA载体的出现为大片段基因敲除提供了更为高效的工具。本研究中使用的PX459- 2sgRNA重组质粒,同时将Cas9基因和双sgRNA转入细胞,与分次转染相比大大提高了外显子切割效率,降低了转染试剂对细胞的毒性。Surveyor法是通过错配酶识别错配的杂合DNA双链并剪切,电泳后将显示出切割条带。sgRNA引导Cas9内切酶在特定位点进行切割后,由于缺乏修复模板,DNA修复主要以NHEJ方式进行,因此将靶序列扩增后经过变性、退火,将形成错配,电泳后切割条带与未切割条带的比例可反映出sgRNA活性。经检测,本研究外显子切割成功率高达50%,而常规方法中单个位点切割也仅可达30%左右[11]。
相比于分离患者肌肉干细胞,iPSC来源广泛,免疫排异反应少,且具有成肌肉干细胞分化潜能和自我更新能力,最有可能为DMD患者提供长期有效治疗[12- 13]。下一步实验中,将利用本研究建立的单质粒CRISPR/Cas9系统敲除DMD基因exon51技术平台,对DMD患者来源的iPSC进行外显子敲除,为DMD的机制和治疗的研究提供实验基础。
参考文献:
[1] Fairclough RJ, Wood MJ, Davies KE. Therapy for duchenne muscular dystrophy: renewed optimism from genetic approaches[J]. Nat Rev Genet, 2013,14:373- 378.
[2] Lu QL, Yokota T, Takeda S,etal. The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy[J]. Mol Ther, 2011,19:9- 15.
[3] Yang J, Li SY, Li YQ,etal. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD[J]. BMC Med Genet, 2013,14:29.
[4] Larson MH, Gilbert LA, Wang X,etal. CRISPR interference (CRISPRi) for sequence-specific control of gene expression[J]. Nat Protoc, 2013,8:2180- 2096.
[5] Mali P, Yang L, Esvelt KM,etal. RNA-guided human genome engineering via Cas9[J]. Science, 2013,339:823- 826.
[6] Cong L, Ran FA, Cox D,etal. Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013,339:819- 823.
[7] Canver MC, Bauer DE, Dass A,etal. Characterization of genomic deletion efficiency mediated by clustered regul-arly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells[J]. J Biol Chem, 2014,289:21312- 21324.
[8] Mendell JR, Campbell K, Rodino-Klapac L,etal. Dystrophin immunity in Duchenne’s muscular dystrophy[J]. N Engl J Med, 2010,363:1429- 1437.
[9] Aartsma-Rus A, Krieg AM. FDA approves eteplirsen for duchenne muscular dystrophy: the next chapter in the eteplirsen saga[J]. Nucleic Acid Ther, 2017,27:1- 3.
[10] Ousterout DG, Perez-Pinera P, Thakore PI,etal. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients[J]. Mol Ther, 2013,21:1718- 1726.
[11] Ousterout DG, Kabadi AM, Thakore PI,etal. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy[J]. Nat Commun, 2015,6:6244. doi:10.1038/ncomms 7244.
[12] Gee P, Xu H, Hotta A. Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy[J]. Stem Cells Int, 2017,2017:8765154. doi: 10.1155/2017/8765154.
[13] Kazuki Y, Hiratsuka M, Takiguchi M,etal. Complete genetic correction of ips cells from Duchenne muscular dystrophy[J]. Mol Ther, 2010,18:386- 393.
