水环境下苯丙氨酸分子旋光异构及羟自由基致损伤的机理
2018-06-13乔朝阳闫红彦杨晓翠王佐成
乔朝阳,高 峰,闫红彦,杨晓翠,佟 华,王佐成
(1.白城师范学院 计算机科学学院,白城 137000; 2.白城师范学院 物理与电子信息学院,白城 137000)
苯丙氨酸(Phe)是一种人体必需氨基酸,分为左旋体(S-Phe)和右旋体(R-Phe).在生命体内S-Phe具有活性,是优构体,还可用作食品添加剂和饲料添加剂.R-Phe可用作制备某种抗癌药物的中间体,用于合成脑内的部分类似肽,还可用于治疗和预防阿尔茨海默病[1].
基于苯丙氨酸的重要作用,人们对它进行了广泛的研究.黄志坚[2]对Phe分子结构进行了比较深入的研究,结果表明,气相Phe分子有多个稳定构象,并计算了几个主要最稳定构象所占的比例和电离能.Simon[3]测量了S-Phe的紫外和红外R2PI谱.Kim小组[4]用密度泛函理论对S-Phe最外壳层轨道的电离能做了研究,研究结果表明,S-Phe的分子内氢键是电离能之间存在差异的原因.Kim[5]用密度泛函理论研究了S-Phe阳离子的构象和能量.Lee[6]通过实验测量了S-Phe的R2PI谱.Pérez等人[7]通过实验测量了S-Phe的转动谱,证实了S-Phe构象存在分子内氢键.张新[8]等采用密度泛函理论的B3LYP方法和微扰理论的MP2方法,对Phe分子的旋光异构裸反应进行了研究,结果表明,基于氨基做质子迁移桥梁是Phe分子旋光异构的优势通道,决速步的能垒是256.7kJ·mol-1.
文献[9-10]的研究表明: 2个水分子簇做质子迁移媒介,对半胱氨酸和亮氨酸分子的质子迁移异构反应具有较好的催化作用,水溶剂有一定的助催化作用.大气中含有大量的水分子并有羟自由基存在,生命体是富水环境,同时也有羟自由基存在.基于此,本工作研究了水汽相和液相环境下,2个水分子簇做质子迁移媒介,Phe分子基于氨基做质子迁移桥梁的旋光异构反应机理以及羟自由基的作用.
1 模型选取与计算方法
采用密度泛函理论的B3LYP[11-12]方法,选用6-31+G(d,p)基组,全优化S-Trp向R-Trp异构过程中2个水分子簇作氢迁移媒介的质子迁移反应各个驻点结构,其他驻点结构取自文献[8].通过对过渡态[13-14]进行内禀反应坐标(IRC)[15-16]计算,验证它们的确是连接所期望的局域极小点.为计算出高水平的势能面,采用微扰理论的MP2方法[17-18],结合6-311++G(2df,pd)基组,计算体系的单点能.计算溶剂效应时,利用气相的构象,采用自洽反应场(SCRF)理论的smd模型方法[19]计算溶剂化的单点能.利用Etotal=ESP+EZPV(ESP为单点能,EZPV为气相的零点振动能)计算总能量.水溶剂环境下,构象1的S型第一中间体与2个水分子簇通过氢键作用形成的络合物记作S-INT1_1·2H2O@water,其他驻点分子表示法相似.计算均由Gaussian09[20]程序完成.
2 结果与讨论
S-Phe分子的2种最稳定构型S-Phe_1和S-Phe_2及手性对映体[8]见图1.研究表明,S-Phe_1和S-Phe_2在水汽相环境下旋光异构过程中,非质子迁移异构反应的机理同于裸反应,在文献[8]已有讨论,这里不再赘述.对于质子迁移过程,水分子簇做质子迁移媒介显著地改变了反应能垒,给予详细地讨论.
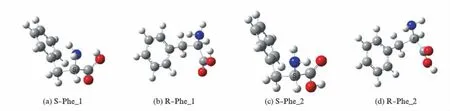
图1 S苯丙氨酸和R苯丙氨酸分子的几何构型Fig.1 The geometries of S and R type phenylalanine molecules
2.1 S-Phe在水环境下的旋光异构
2.1.1 S-Phe_1在水汽环境下的旋光异构及水溶剂效应
在水汽环境下,2个水分子簇作氢迁移媒介S-Phe_1的旋光异构历程见图2,在水汽相和液相反应过程的势能面见图3(看第208页).
第1基元反应是羧基从反式平面结构向顺式平面结构的异构.23H从羧基左侧转到右侧,同时,2H和21H顺时针旋转,氨基氮电荷密度大的一面(有孤对电子)朝向读者,这为水分子簇在S-Phe_1的正面与S-Phe_1通过氢键作用形成络合物分子创造了条件.此基元反应过程几何参数的变化在文献[8]已做了详细讨论,不再赘述.
第2基元反应是水分子簇催化的质子在纸面外从手性碳向氨基氮迁移.首先S-INT1_1与手性碳3C上的23H和氨基氮1N前面的2个水分子簇通过氢键作用形成前驱络合物S-INT1_1·2H2O(m)(m表示水分子在S-INT1_1的前面,图2给出的是右视图),氢键能是65.56kJ·mol-1.S-INT1_1·2H2O(m)经络合物过渡态TS2_1·2H2O(m)异构成中间体络合物INT2_1·2H2O(m),实现了质子从手性碳向氨基氮的净迁移.过渡态TS2_1·2H2O(m)的七元环结构中,氢键键角3C-23H-24O、24O-27H-28O和28O-25H-1N分别是161.44°、160.82°和162.18°,均接近平角,3个氢键均比较强;二面角3C-23H-24O-27H、24O-27H-28O-25H和28O-25H-1N-3C分别是-29.12°、-7.60°和-35.43°,七元环结构接近平角,因此TS2·2H2O(m)比较稳定.又从S-INT1·2H2O(m)到TS2_1·2H2O(m)过程,反应活性中心骨架二面角1N-3C-4C-5C从129.83°变为129.02°,骨架没有形变.因此,TS2_1·2H2O(m)产生的能垒不会太高.但从S-INT1_1·2H2O(m)到TS2_1·2H2O(m)的过程中,3C-23H、24O-27H和28O-25H分别从0.10995、0.09805和0.09943nm增加到0.13155、0.14631和0.16175nm,3个化学键较大幅度的拉伸断裂还是需要一定的能量的.因此,TS2_1·2H2O(m)产生了123.98kJ·mol-1的能垒,这比裸反应的能垒256.70kJ·mol-1[8]降低了51.7%,说明2个水分子簇起到了较好的催化作用.中间体产物络合物INT2_1·2H2O(m)是质子从手性碳向氨基氮净迁移的产物,质子从手性碳向氨基氮净迁移导致体系正负电荷分离,INT2_1·2H2O(m)体系不稳定,处在势能面上相对能量较高的位置.中间体产物络合物INT2_1·2H2O(m)脱去水分子,需要克服的氢键能是100.35kJ·mol-1.
第3基元反应是水分子簇催化的质子从氨基氮在纸面里向手性碳迁移.首先,INT2_1与纸面里侧2C和23H对面的2个水分子簇通过氢键作用形成氢键络合物INT2_1·2H2O(n)(n表示水分子簇在纸面里与底物络合,图2给出的是右视图),氢键能是85.83kJ·mol-1.然后,INT2_1·2H2O(n)经过渡态络合物TS3_1·2H2O(n),实现了质子23H从氨基氮1N向手性碳2C(原来序号是3C)的净迁移,异构成R型中间体络合物R-INT3_1·2H2O(n),完成旋光异构.
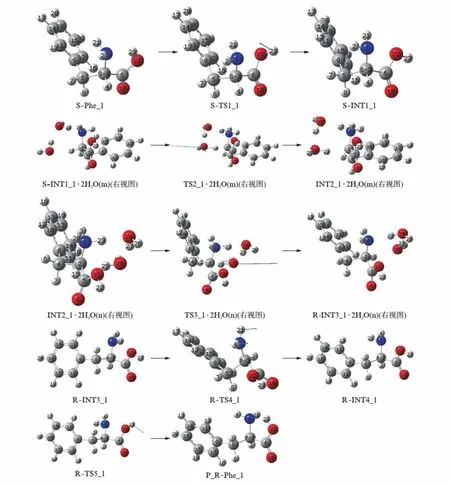
图2 水汽环境下S-Phe_1旋光异构的反应历程Fig.2 The reaction process of S-Phe_1 optical isomerism in water vapour
过渡态TS3_1·2H2O(n)的七元环结构的氢键角2C-25H-28O、28O-27H-24O和24O-23H-1N分别是163.35°、160.79°和161.16°,接近平角,3个氢键较强;二面角2C-25H-28O-27H和28O-27H-24O-23H分别是-8.38°和-6.51°,接近平角.因此,七元环结构过渡态TS3_1·2H2O(n)比较稳定,不会产生太高的能垒.从INT2_1·2H2O(n)到TS3_1·2H2O(n)过程,1N-23H、24O-27H和28O-25H分别从0.10497、0.09901和0.10057nm增加到0.10761、0.10436和0.12836nm,增幅远远小于从S-INT1_1·2H2O(m)到TS2_1·2H2O(m)过程相应的化学键的增幅.因此,TS3_1·2H2O(n)产生的能垒较低,只有11.61kJ·mol-1.中间体产物络合物R-INT3_1·2H2O(n)是质子从氨基氮向手性碳净迁移的产物,质子从氨基氮向手性碳净迁移拉近了体系正负电荷中心的距离,使体系变得稳定,再加上R-INT3_1·2H2O(n)体系存在3个分子内氢键和氨基与苯环间的π氢键,导致R-INT3_1·2H2O(n)体系处在势能面上相对能量较低的位置(-205.29kJ·mol-1).此基元反应是放热反应,所放热量导致R-INT3_1和H2O分子间的氢键轻而易举地断裂.气相反应的能垒一般采用总包能垒(表观能垒),从图3可以看出,S-Phe_1在水汽环境下的旋光异构的总包能垒(表观能垒)为145.27kJ·mol-1,已经远高于温和反应的“能垒”84.01kJ·mol-1[21],说明水汽相环境下S-Phe_1分子很难消旋.
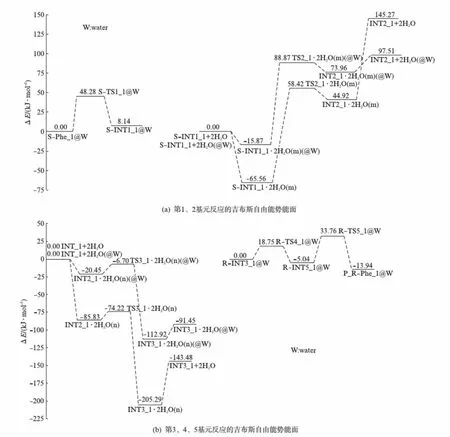
图3 S-Phe_1旋光异构反应的吉布斯自由能势能面Fig.3 Gibbs free potential energy surfaces diagram of the optical isomerism reaction of S-Phe_1
比较图3的2水分子簇助S-Phe_1旋光异构过程中的质子迁移反应和文献[8]的图5可知,2个水分子簇将决速步能垒从256.70kJ·mol-1降到124.28kJ·mol-1,使第3基元反应能垒从117.70kJ·mol-1降到11.61kJ·mol-1,说明水分子簇对S-Phe_1旋光异构过程中的质子迁移反应有着较好的催化作用.
从图3还可以看出: 水溶剂效应使水分子簇催化S-Phe_1旋光异构过程中的决速步质子迁移反应能垒进一步降到104.74kJ·mol-1,说明水溶剂效应对S-Phe_1旋光异构过程中的水助质子迁移反应有着一定的助催化作用.104.74kJ·mol-1已经远远低于质子迁移的“极限能垒”167.00kJ·mol-1[21]许多,说明水液相环境下S-Phe_1可以缓慢地消旋化.
比较图3和文献[8]的图5还可以看出: 水溶剂效应使羧基异构的能垒有所降低,使氨基异构的能垒略有升高.
2.1.2 S-Phe_2在水汽环境下的旋光异构及水溶剂效应
在水汽环境下,2个水分子簇作氢迁移媒介S-Phe_2的旋光异构历程见图4,在水汽相和液相反应过程的势能面见图5,各基元反应物和产物氢键络合物的形成与解离机理同于S-Phe_2的情形,这里从略,仅讨论2个水分子簇作氢迁移媒介的质子迁移过程.
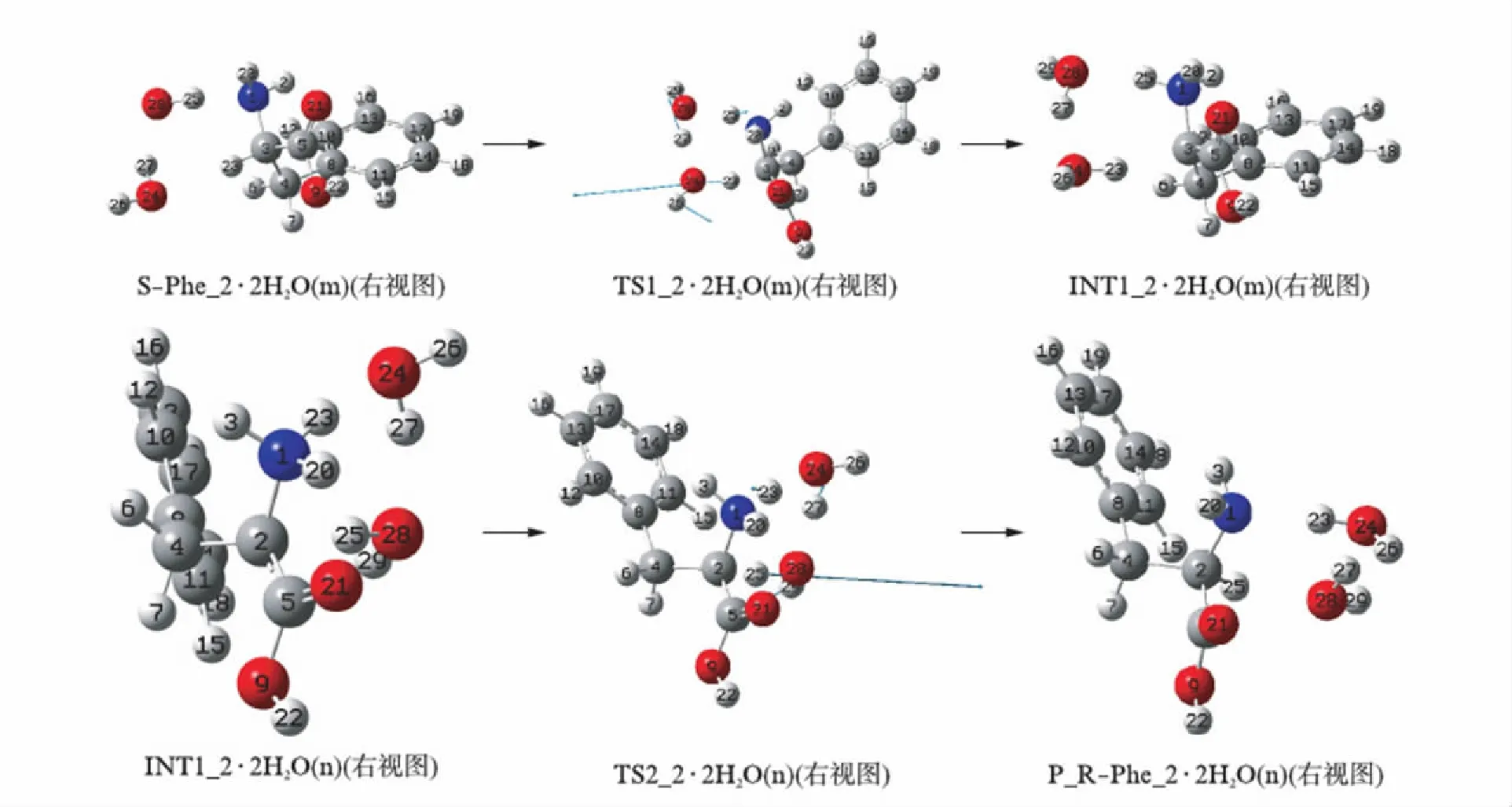
图4 S-Phe_2旋光异构的反应历程Fig.4 The reaction process of S-Phe_2 optical isomerism

图5 S-Phe_2旋光异构反应的吉布斯自由能势能面Fig.5 Gibbs free potential energy surfaces diagram of the optical isomerism reaction of S-Phe_2
首先S-Phe_2与23H和1N前面的2个水分子簇通过氢键作用形成的络合物S-Phe_2·2H2O(m)(m表示水分子在S-INT1_1的前面,图4给出的是右视图),经络合物过渡态TS1_2·2H2O(m)异构成中间体络合物INT1_2·2H2O(m),实现了质子从手性碳3C向氨基氮1N的净迁移.结构分析表明: 此基元反应同于2.1.1中的第2基元反应,过渡态TS1_2·2H2O(m)的七元环结构相似于TS2_1·2H2O(m),3个氢键均均接近平角,氢键较强;七元环结构的二面角接近平角.因此,TS1_2·2H2O(m)比较稳定.再加上从S-Phe_2到TS1_2·2H2O(m)过程,反应活性中心骨架基本没有形变.因此,TS1_2·2H2O(m)产生的能垒也不会太高.但从S-Phe_2到TS1_2·2H2O(m)过程同于S-INT1_1·2H2O(m)到TS2_·2H2O(m)过程,3C-23H、24O-27H和28O-25H这3个化学键也有较大幅度的拉伸断裂,需要一定的能量.因此,TS1_2·2H2O(m)产生了119.85kJ·mol-1的能垒,这比裸反应的能垒256.50kJ·mol-1[8]大幅下降,说明2个水分子簇起到了较好的催化作用.中间体产物络合物INT1_2·2H2O(m)相似于INT2_1·2H2O(m),其正负电荷中心分离,导致体系不稳定,也处在势能面上能量较高的位置.
然后,INT1_2与纸面里侧2C和23H对面的2个水分子簇通过氢键作用形成氢键络合物INT1_2·2H2O(n),经过渡态络合物TS2_2·2H2O(n),实现了质子23H从氨基氮1N向手性碳2C(原来序号是3C)的净迁移,异构成R型产物络合物P_R-Phe_2·2H2O(n),完成旋光异构.结构分析表明,此基元反应同于2.1.1中的第3基元反应,过渡态TS2_2·2H2O(n)七元环结构的3个氢键角接近平角,3个氢键都较强;二面角2C-25H-28O-27H和28O-27H-24O-23H也接近平角.因此,TS2_2·2H2O(n)比较稳定,也不会产生太高的能垒.从INT1_2·2H2O(n)到TS2_2·2H2O(n)的过程中,1N-23H、24O-27H和28O-25H增幅很小,所需能量不是很高.因此,TS2_2·2H2O(n)产生的能垒很低,只有17.75kJ·mol-1.产物络合物P_R-Phe_2·2H2O(n)是质子从氨基氮向手性碳净迁移的产物,质子从氨基氮向手性碳净迁移拉近了体系正负电荷中心的距离,使体系变得稳定,再加上P_R-Phe_2·2H2O(n)体系存在3个分子内氢键和氨基与苯环间的π氢键,导致P_R-Phe_2·2H2O(n)体系处在势能面上较低的位置(-112.53kJ·mol-1),此基元反应是放热反应.
2.2 水汽环境下羟自由基致S-Phe损伤及水溶剂效应
研究表明,羟自由基本身并不会致S-Phe损伤,需有一个水分子与羟自由基通过氢键作用构成链,水分子羟自由基链与Phe分子通过氢键作用形成的络合物异构导致Phe分子损伤.水汽环境下羟自由基致S-Phe_1和S-Phe_2损伤的机理稍有不同,S-Phe_1是羧基异构后的中间体损伤,S-Phe_2是直接损伤,下面分别进行讨论.
2.2.1 水汽环境下羟自由基致S-Phe_1损伤及水溶剂效应
由于S-Phe_1的22H对水分子羟自由基链在23H和1N的前面与其形成氢键有空间位阻,不能直接在23H和1N的前面与水分子羟自由基链形成氢键,羧基从反式平面结构向顺式平面结构异构后,中间体产物S-INT1_1与水分子羟自由基链通过氢键作用,形成了前驱络合物,S-INT1_1损伤了也就是S-Phe_1损伤了.水分子羟自由基链与S-INT1_1形成氢键有两种情况,一种是羟自由基与氨基氮形成氢键,水分子与手性碳上的氢形成氢键,S-INT1_1损伤是水分子拔氢机理;另一种是水分子与氨基氮形成氢键,羟自由基与手性碳上的氢形成氢键,S-INT1_1损伤是羟自由基拔氢机理,水分子本身并没有化学键的断裂与生成,它是通过与S-INT1_1和羟自由基形成氢键相互作用才起到了催化作用,它的另一个作用是稳定羟自由基的空间位置.S-INT1_1损伤的反应历程见图6的(a)与(b),反应势能面见图7(a)(看第212页).
第一种情况形成的前驱络合物记作S-INT1_1·(H2O·OH·),其经过渡态络合物TS2_1·(H2O·OH·),实现了27H向羟基的迁移和23H向24O的迁移,形成了产物络合物,记作P*a_1·2H2O.Pa_1的手性已不存在,其已不具有苯丙氨酸的特性,即S-INT1_1损伤(S-Phe_1损伤了).该基元反应的产物是P*a_1·2H2O,并不是氨基质子化的INT2_1·(H2O·OH·)(m).这是由于羟自由基有着很强的夺取质子的能力,把迁移到氨基上的质子又夺了回来.对过渡态TS2_1·(H2O·OH·)进行的IRC计算表明,此基元反应是三氢迁移机理,过渡态之后水分子23H-26H-24O和羟自由基27H-28O形成,质子化氨基即将形成;由于质子化氨基不稳定,容易失去一个质子,羟自由基27H-28O夺取了质子化氨基上的25H,形成了水分子27H-28O-25H;最后,两个水分子分别与羧基和苯环形成氢键,两个水分子间形成氢键,氨基和苯环之间形成了π氢键,骨架原子1N-3C-4C-5C-9O-21O形成了稳定的大π键.因此,得到了十分稳定的产物络合物P*a_1·2H2O(P*表示氨基酸损伤的产物,下同),其相对于S-INT1_1·(H2O·OH·)的能量是-178.93kJ·mol-1.
过渡态TS2_1·(H2O·OH·)七元环结构的氢键角3C-23H-24O、24O-27H-28O和28O-25H-1N分别是160.02°、159.26°和165.48°,均接近平角,3个氢键均比较强;二面角3C-23H-24O-27H、24O-27H-28O-25H和27H-28O-25H-1N分别是-25.74°、1.58°和-38.43°,七元环结构接近平角,TS2_1·(H2O·OH·)较稳定.又从S-INT1_1·(H2O·OH·)到TS2_1·(H2O·OH·)的过程中,反应活性中心骨架二面角1N-3C-4C-5C从129.59°变为129.41°,骨架基本没形变.因此,TS2_1·2H2O(m)产生的能垒不会太高.但从S-INT1_1·(H2O·OH·)到TS2_1·(H2O·OH·)的过程中,3C-23H、24O-27H和28O-25H分别从0.10988、0.09773和0.10186nm增加到0.13585、0.14667和0.17292nm,3个化学键较大幅度的拉伸断裂是需要一定的能量的.TS2_1·(H2O·OH·)产生的能垒是124.97kJ·mol-1.
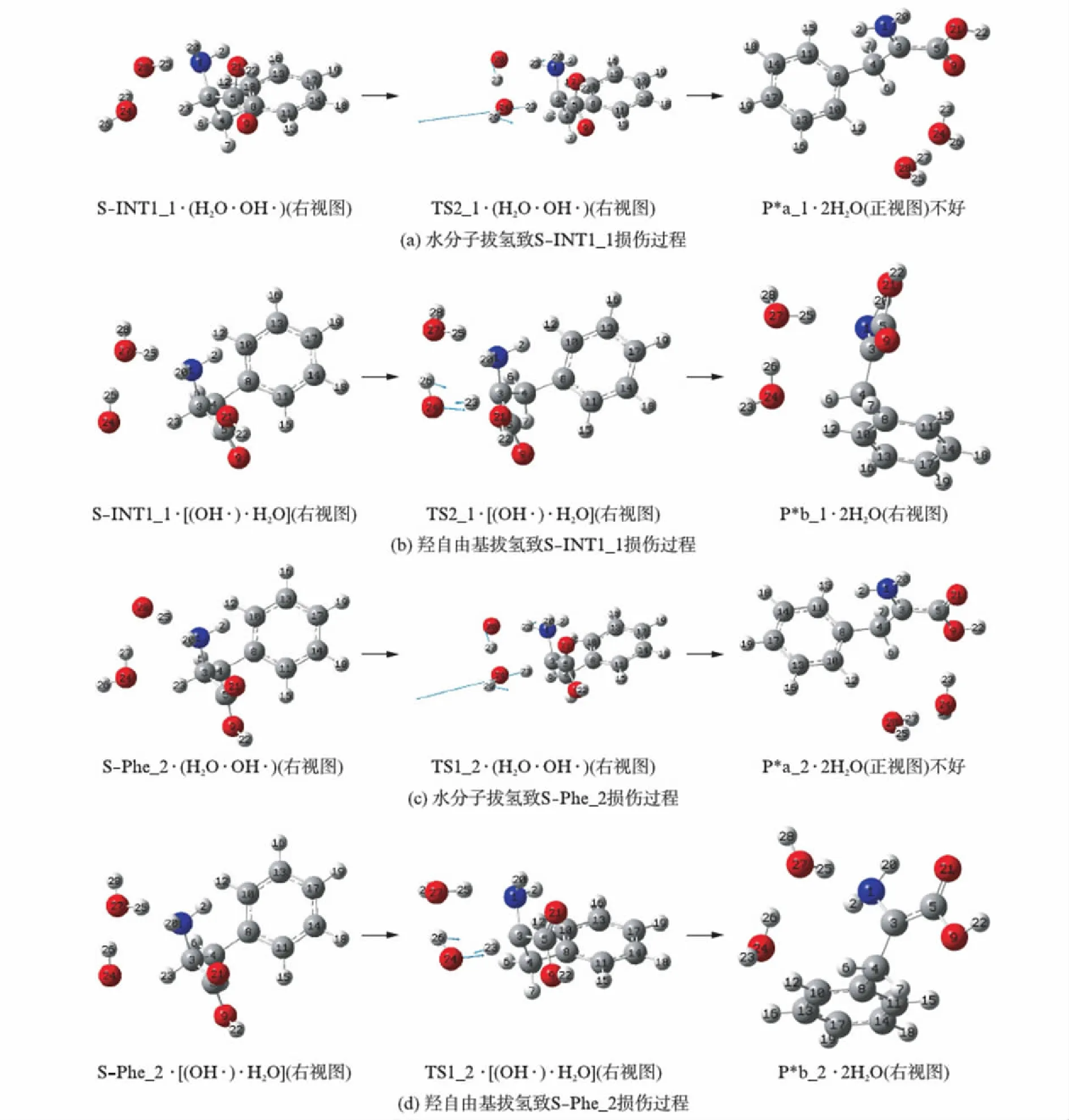
图6 水分子羟自由基链致苯丙氨酸分子损伤的过程Fig.6 The phenylalanine molecules damage process introduced by hydroxyl free radical of water
从图7(a)可以看出: 溶剂效应使水分子和羟自由基作氢迁移媒介,水分子拔手性碳的氢致S-Phe_1损伤的能垒降为28.33kJ·mol-1,此基元反应放热339.66kJ·mol-1.28.33kJ·mol-1已经远低于温和反应的能垒84.01kJ·mol-1[21],这说明溶剂效应使水分子和羟自由基作氢迁移媒介,水分子拔氢致S-Phe_1损伤过程可以在温和条件下进行.
第二种情况形成的前驱络合物记作S-INT1_1·[(OH·)·H2O],经过渡态络合物TS2_1·[(OH·)·H2O],实现了氢原子23H从手性碳向24O的迁移,形成了产物络合物,记作P*b_1·2H2O.此过程是单氢转移机理,羟自由基夺取了手性碳上的氢原子,形成了水分子.P*b_1的手性也已不存在,已不具有苯丙氨酸的特性,即S-INT1_1损伤(S-Phe_1损伤了).结构分析表明,从S-INT1_1·[(OH·)·H2O]到TS2_1·[(OH·)·H2O]的过程中,只是3C-23H键长从0.10996nm拉伸到0.11536nm,拉伸幅度是0.00540nm,其他结构参数变化甚微.3C-23H键长如此小的拉伸所需能量很小,因此,TS2_1·[(OH·)·H2O]产生的能垒很小,只有21.06kJ·mol-1.21.06kJ·mol-1已经远低于温和反应的能垒84.01kJ·mol-1[23],水分子羟自由基链可以致S-INT1_1比较快地损伤(即S-Phe_1迅速损伤).结构分析表明,P*b_1·2H2O的1N-3C-4C-5C-9O-21O形成了稳定的大π键,又具有水分子之间的氢键、水分子与氨基之间较强的单氢键、氨基与苯环间的π氢键以及水分子与苯环之间的单氢键.因此,它是十分稳定的产物络合物,相对于S-INT1_1·[(OH·)·H2O]的能量是-159.39kJ·mol-1.
从图7可以看出: 溶剂效应使羟自由基夺氢原子致S-Phe_1损伤的能垒降为-113.05kJ·mol-1,产物的相对能量是-307.92kJ·mol-1.即水溶剂效应使该情形的S-Phe_1损伤变成了无势垒放热反应,也就是说水溶剂相羟自由基夺取手性碳上的氢原子致S-Phe_1损伤过程可以极快的速度进行.
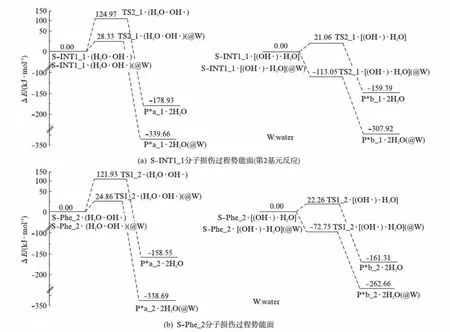
图7 水分子和羟自由基链致苯丙氨酸分子损伤反应过程势能面Fig.7 Potential energy surfaces diagram of the phenylalanine molecules damage reaction introduced by hydroxyl free radical and water
2.2.2 水汽环境下羟自由基致S-Phe_2损伤及水溶剂效应
S-Phe_2可直接与其23H和1N前面的羟自由基水分子链形成氢键,S-Phe_2损伤也存在羟自由基拔氢的单氢转移和水分子拔氢的三氢转移两个通道的竞争,相似于S-INT1_1的损伤,不再赘述.
水分子拔氢时损伤能垒是121.93kJ·mol-1,羟自由基拔氢时损伤能垒是22.26kJ·mol-1,22.26kJ·mol-1已远远低于温和反应的能垒84.01kJ·mol-1[21],因此,水汽环境下羟自由基可致S-Phe_2损伤.溶剂效应使这两个能垒分别降为24.86kJ·mol-1和-72.75kJ·mol-1.这说明水汽相环境下是羟自由基拔氢致S-Phe_2损伤,水溶剂环境下是羟自由基拔氢和水分子拔氢致S-Phe_2损伤两种机理共存,只是后者更具优势.
从图7可以看出,水分子和羟自由基链致S-INT1_1(也就是S-Phe_1)和S-Phe_2分子损伤反应,对于羟自由基拔氢的单氢转移和水分子拔氢的三氢转移两个通道的竞争,无论是在水汽相还是在水溶剂相,都是前者占有绝对优势.水溶剂对Phe分子损伤反应具有极好的助催化作用.
2个水分子簇的催化使构型1旋光异构决速步能垒降到123.98kJ·mol-1,使构型2旋光异构决速步能垒降到119.85kJ·mol-1.水溶剂效应使这两个能垒进一步降到104.74和103.68kJ·mol-1.水分子和羟自由基链致Phe分子损伤过程,水分子拔氢致Phe_1和Phe_2损伤的能垒是124.97和121.93kJ·mol-1,水溶剂效应使这两个能垒分别降到28.33和24.86kJ·mol-1;羟自由基拔氢致Phe_1和Phe_2损伤的能垒是21.06和22.26kJ·mol-1,水溶剂效应使两个能垒分别降到-113.05和-72.75kJ·mol-1.结果表明: 水分子簇对Phe分子的旋光异构具有较好的催化作用,水溶剂具有助催化作用;水分子羟自由基链可导致Phe分子损伤,水溶剂对损伤过程有着极好的助催化作用,羟自由基拔氢的单氢转移通道更具优势.
参考文献:
[1] 方岩雄,张维刚,杨顺利,等.苯丙氨酸的应用与生产 [J].广州化工,2000,28(4): 134-135.
[2] 黄志坚.氨基酸的构型和性质研究 [D].合肥: 中国科学技术大学,2006: 11.
[3] SNOEK L C, ROBERTSON E G, KROEMER R T, et al. Conformational landscape in amino acids: Infrared and ultaaviolet ion-dip spectroscopy of phenylala-nine in the gas phase [J].ChemPhysLett, 2000,321(1/2): 49.
[4] LEE K T, SUNG J, LEE K J, et al. Conformation-dependent ionization energies of L-phenylalanine [J].AngewChem, 2002,114(21): 4288.
[5] LEE K T, SUNG J, LEE K J, et al. Conformation-dependent ionization of L-phenylalanine: Structures and energetics of cationic conformers [J].ChemPhysLett, 2003,368(3/4): 262.
[6] LEE Y, JUN J, KIM B, et al. Alanyl side chainfolding in phenylalanine: Conformational assignments through ultraviolet rotational assignments through ultraviolet rotational band contouranallysis [J].JPhysChemA, 2004,108(1): 69.
[7] PÉREZ C, MATA S, BLANCO S, et al. Jet-cooled rota-tional spectrum of laser-ablated phenylalanine [J].PhysChemA, 2011,115(34): 9653.
[8] 张新,李晨洁,董丽荣,等.基于氨基做质子迁移桥梁苯丙氨酸分子的旋光异构反应机理 [J].复旦学报(自然科学版),2017,56(2): 241-250.
[9] 王佐成,范志琳,梅泽民,等.半胱氨酸分子手性转变及水分子的催化机理 [J].武汉大学学报(理学版),2016,62(4): 368-374.
[10] 赵晓波,李晨洁,高峰,等.基于氨基做质子迁移桥梁亮氨酸的手性转变机理及水溶剂化效应 [J].中山大学学报(自然科学版),2017,56(3): 85-92.
[11] BECKE A D. Density-functional thermochemistry. Ⅲ. The role of exact exchange [J].ChemPhys, 1993,98(7): 5648-5652.
[12] PARR R G, YANG W. Density-functional theory of atoms and molecules [M]. Oxford,UK: Oxford University Press, 1994.
[13] GARRETT B C, TRUHLAR D G. Generalized transition state theory. Classical mechanical theory and applications to collinear reactions of hydrogen molecules [J].JPhysChem, 1979,83(8): 1052-1079.
[14] GARRETT B C, TRUHLAR D G. Criterion of minimum state density in the transition state theory of bimolecular reactions [J].JChemPhys, 1979,70(4): 1593-1598.
[15] GONZALEZ C, SCHLEGEL H. Reaction path following in mass-weighted internal coordinates [J].JPhysChem, 1990,94(14): 5523-5527.
[16] ISHIDA K, MOROKUMA K, KOMORNICKI A. The intrinsic reaction coordinate. An ab initio calculation for HNC→HCN and H-+ CH4→CH4+ H-[J].JChemPhys, 1977,66(5): 2153-2156.
[17] 徐光宪,黎乐民,王德民.量子化学(中册) [M].北京: 科学技术出版社,1985: 962-986.
[18] BINKLEY J S, POPLE J A. Moeller-Plesset theory for atomic ground state energies [J].IntJQuantumChem, 1975,9(2): 229-236.
[19] ALEKSANDR V M, CHRISTOPHER J C, DONALD G T. Universal slovation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions [J].JPhysChemB, 2009,113(18): 6378-6396.
[20] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09. Revision E.01 [CP]. Pittsburgh USA: Gaussian, Inc., Wallingford CT, 2013.
[21] GORB L, LESZCZYNSKI J. Intramolecular proton transfer in mono- and dihydrated tautomers of guanine: An ab initio post hartree-fock study [J].JAmChemSoc, 1998,120(20): 5024-5032.
