宏基因组大数据分析的质量控制流程规范
2018-06-01郑广勇杨桢曹瑞芳刘婉李亦学张国庆
郑广勇,杨桢,曹瑞芳,刘婉,李亦学,,张国庆,
1.中国科学院上海生命科学研究院生物医学大数据中心, 上海 200031;
2.上海生物信息技术研究中心, 上海 201203
1 引言
近年来,随着二代测序技术及各种高通量组学技术的快速发展,生物医学研究进入大数据时代。实验技术和信息技术的发展,使生物医学数据开始从万亿字节的TB级跃升到千万亿字节的EB级,引发了后基因组时代的生物医学研究的深刻变革。生物医学研究已经从第一范式的实验科学,或快或慢地进入第二范式理论科学、第三范式计算科学,直到最新的第四范式数据密集型科学。数据质量对大数据分析与挖掘的价值日益凸显。目前,测序能力的提升极大地推动了包括微生物组学在内的各种组学的快速发展,进而催生了大量以微生物为主要研究对象的国际合作项目,使得微生物组和微生物系统组成为生物医学大数据的重点研究方向之一。微生物组是指存在于特定环境或生态系统中的微生物以及它们携带的遗传信息和生物学功能的总和[1]。微生物组与人类健康有极为重要的关系,人体微生物组由数百至数千种不同的微生物组成,其细胞总数可达数万亿之多,数量远超人体自身细胞数量,其中独特的微生物基因数量多达2 000万个,数目远远超过人的基因数目(大约 2.5 万个)。通过对人体微生物组进行研究,可以解析人类健康、营养、代谢等方面的科学问题。
2 国内外相关研究
目前,宏基因组(metagenome)技术是微生物组研究的重要手段之一,该技术利用基因组学策略研究特定环境样品中包含的全部微生物的遗传组成及其功能模式[2]。宏基因组技术直接从环境样品中提取DNA样本,避开了传统的微生物分离培养方法,为研究和利用占微生物种类99%以上的不可培养的微生物提供了一种新的途径和良好的策略。近年来,高通量测序技术的快速发展为微生物表型及其遗传机制的探索提供了新的技术方案[3]。宏基因组技术在农业、林业、环保、医药等领域都有着广泛的应用,提供了丰富的人体、动植物、各类环境的科学研究数据[4-8]。2005年以来,以国际宏基因组联盟为代表的微生物组研究计划带动了很多国家的相关研究,例如美国的人类微生物组计划(Human Microbiome Project,HMP)[9]和欧盟的人类肠道微生物联盟(Metagenomics of the Human Intestinal Tract Consortium,MetaHIT)[10]。这些项目的实施,推动了数据库和数据挖掘等基础研究工作的开展,并对相关数据标准与质量控制等提出了新的要求。相对传统组学数据而言,宏基因组测序数据量大、涉及的微生物基因组复杂度高,而且可能存在大量未知物种,传统的基于单一组学或单一物种的分析方法无法直接应用于宏基因组研究,针对宏基因组数据的采集、整理、存储以及后续研究分析等仍缺乏统一的规范。因此,宏基因组研究急需建立一套涵盖样本信息以及测序数据采集、整理、存储、交换、分析的数据规范与标准。从数据流程看,宏基因组的分析过程包括元数据和测序数据质量控制、微生物群落组成分析、群落功能生态分析、菌群差异功能分析等步骤(如图1所示)。其中,元数据和测序数据质量控制是后续一系列分析的基础,直接影响整个分析的完整性和正确性,因而在整个数据分析过程中具有十分重要的意义。本文对宏基因组研究中的元数据及测序数据的预处理过程中的相关质量控制标准进行了简要探讨,从而为宏基因组数据分析提供支撑。
3 宏基因组数据质量控制
宏基因组的测序数据具有数据量大、复杂度高的特点,其数据分析有一些特定的要求,具体而言主要有以下几个方面。
● 宏基因组研究中样本收集、存储、运输等信息都对微生物菌群差异分析有重要影响,因此需要在元数据中记录这些信息,并在质量控制流程中对元数据和测序数据进行一致性检查,以保证后续分析的正确性。
● 宏基因组研究通常依赖二代或三代测序技术,其测序速度比一代测序技术有显著提升,但其测序长度及准确度却有所下降,因此在宏基因组数据质量控制流程中必须对数据进行去噪、剔除接头、切除无效序列等操作,以保证结果的可靠性。
● 与宿主相关的微生物样品测序结果中通常包含一定数量的宿主基因片段,这些片段会对后续的微生物菌群分析产生干扰,因此在宏基因组数据质量控制流程中必须去除这些宿主基因片段,以保证后续分析的有效性。
● 需要对样本的测序深度进行一定的评估,从而保证不同分组样本差异分析的需求。
笔者在长期的微生物组大数据分析过程中,根据宏基因组的数据特点和数据分析的要求,提出了一套完整的数据质量控制流程(如图2所示),具体包括:元数据和测序数据的一致性检查、测序数据的质量检测、低质量测序序列的过滤及切除、接头序列及无关序列的剔除、宿主及污染序列的过滤、混合样本的数据分割、样本菌群的组成分析、不同分组样本的主坐标分析(principal coordinateanalysis,PCoA)这8个步骤,下面将对这些步骤进行详细论述。

图1 宏基因组数据主要分析流程

图2 宏基因组数据质量控制流程
3.1 元数据和测序数据的一致性检查
元数据是对数据进行描述的数据(data about data),主要对数据的属性进行表述。元数据主要包括数据来源、数据收集整理模式以及数据可靠性等信息,是实验数据的重要组成部分。就宏基因组研究而言,元数据提供了项目技术设计、材料来源、实验方案、结果描述等至关重要的信息[11]。目前国际基因组标准联盟(Genomic Standards Consortium,GSC)已开发了针对基因组[12]、宏基因组[13]及扩增子[14]研究的多种元数据标准框架,框架包括组学数据的检查列表以及取样环境描述包(environmental package)。其中,宏基因组数据标准框架的制定原则指出,人体样本和环境样本的最少信息集合应当以样本为出发点,保证样本的基因型和表型关联分析、不同分组样本的差异分析、样本菌群组成的机理研究分析的需求,因此笔者建议宏基因组的元数据中应当包含如下基本信息。
● 人体样本:个人基本生理信息、生活行为方式、膳食结构、营养状况、既往病史等信息。
● 环境样本:样本获取过程中的采集地点、大气、水文、温度、压力、运输方法、存储媒介等信息。
同时,笔者建议使用国际宏基因组数据标准框架制定的标准词汇来描述元数据,从而给出精确的元数据语义信息,进而建立可共享的、可被机器处理的本体支持,利于未来的数据资源整合。在元数据完整性检查完成后,需要开展元数据与测序数据的一致性检查,重点检查测序数据的样本是否符合元数据描述的样本,查看是否存在数据遗漏、差错匹配、错误标注等现象,这些都会对后续的数据分析造成严重的影响。这种一致性检查是数据完整性、有效性的重要质量控制步骤。
3.2 测序数据的质量检测
目前,宏基因组研究大都采用二代测序技术。以illumina为代表的二代测序技术基本都运用边合成边测序的策略,在碱基链合成的过程中,随着合成链的增长,DNA聚合酶的效率会不断下降,特异性也逐渐变差,从而造成碱基合成错误率增高。此外,测序仪在开始进行合成反应时,也会由于反应不够稳定带来质量值的波动。测序数据的质量好坏会影响下游的分析,不同测序平台的测序错误率存在较大差异,因此在数据分析前,需要确定原始数据是通过哪种测序平台产生的、其错误分布规律如何、是否存在一定测序偏向性、是否受序列中碱基含量分布(GC含量)影响等[15]。目前对原始测序数据进行质量检测,通常使用FastQC和PRINSEQ[16]这两种检测软件包。FastQC软件包既有图形操作界面,又有命令行操作模式,能同时支持多种文件格式,包括FastQ、SAM和BAM格式等。其测序质量报告主要包括:读段(reads)各位置的碱基质量值分布、碱基的总体质量值分布、reads各个位置上碱基分布比例、GC含量分布、reads各个位置的非确定碱基数目、是否含有测序接头序列等。PRINSEQ是另一款常用的测序质量检测软件,其报告内容主要包括:reads数目以及长度频率分布、碱基质量频率分布、序列复杂度、GC含量、重复序列及接头信息等。
3.3 低质量测序序列的过滤及切除
原始测序数据通常包含测序分值较低的碱基序列,其处理方式包括过滤和切除。对于全长质量都比较低的序列,可直接进行过滤处理;而对于只有部分片段质量较低的序列,则可以通过片段切除的方法来处理。最基本的切除方法为设定特定的切除长度或剩余长度,这种方法会去除部分测序质量较好的片段,从而造成一定的信息损失,因此更为通用的方案是从序列任意一端开始,逐个切除低于质量阈值的碱基。如目前较为流行的低质量序列切除与过滤软件Trimmomatic[17]及PRINSEQ均支持从任意一端开始切除低质量碱基。另一种方案为滑窗策略,将窗口内的碱基质量与设定的阈值进行比较,如果滑窗内的碱基数值低于质量阈值,则切除整个滑窗的碱基。用户可根据实际情况设置滑窗大小及质量阈值。另外,值得注意的是,测序时如果无法判定一个碱基是哪种碱基,通常标记为N字符,不同的组装比对软件对于N字符的处理方式完全不同,有的用4种碱基随机替代N字符,有的则是用固定的某个碱基替代N字符,由于N字符会导致错误的组装和比对,因此含N字符较多的序列同样需要在组装和比对前进行切除或过滤操作。
3.4 接头序列及无关序列的剔除
高通量测序过程一般会使用测序接头序列,接头序列的存在可能会导致基因组组装和转录本组装出现问题,因此需要在分析数据之前予以去除。此外,其他的测序标签以及引物片段也需要去除。去除接头及标签序列是一项比较困难的任务,首先,这些序列可能存在测序错误,需要考虑应对错配、插入缺失片段(indels)以及不确定碱基(N字符)的情况;其次,如果测序的目标序列较短,reads可能会延伸到3’端的接头序列。而这种“读穿”的情况会导致reads中含有部分3’端的接头序列无法被识别。此外,某些公共来源的测序数据可能根本无法知道接头序列信息。目前较为常用的接头序列切除软件包括Trimmomatic[17]、TagCleaner[18]和Cutadapt等。这些软件均可以应对错配,并允许用户指定测序和标签序列的最小重叠,TagCleaner还可以应对indels以及不确定碱基。假如接头序列未知,可以先用TagCleaner软件预测,再进行后续的切除。另外,值得注意的是,在测序文库的制备过程中,聚合酶链式反应(PCR)过度扩增可能导致重复序列的出现,在后续分析过程中,这些重复序列应当剔除。此外,某些低复杂度的序列由于缺乏信息量,很难可靠地比对到参考序列上,因此也应当予以剔除。
3.5 宿主及污染序列的过滤
宏基因组测序是对样本中所有DNA分子进行测序,因此获得的数据中可能含有不属于微生物的序列,例如与人类健康相关的宏基因组研究多使用与人体相关的组织或样本(如口腔菌斑、唾液、皮肤及粪便样本等),这些样本中可能存在人类基因组序列污染。此外,研究样本也可能存在其他生物体或载体的序列污染,从而对后续分析造成影响,因此在质量控制过程中需要剔除宿主序列以及可能的污染序列。最直接的方法就是把序列比对至宿主基因组及可能的污染源序列上,然后剔除这些序列。FastQ Screen、BWA[19]、Bowtie[20]、SOAP等工具可以把序列比对至用户怀疑的污染源序列上,如果存在一致序列则予以剔除。
3.6 混合样本的数据分割
测序时,为了区别不同样本来源的序列,需要在待测序列中加入一段具有特定序列的编码序列(barcode)。在质量控制过程中,为了获得不同样本的完整数据,需要根据码序列对混合样本进行分割,将一个FastQ文件中的序列分别存储到多个样本文件中。
3.7 样本菌群的组成分析
在获得测序数据之后,首要工作是对相关样本进行菌群组成分析。在早期宏基因组研究中,通常需要对测序数据进行拼接,获得contig序列,之后通过一些常用的序列比对算法(如BLAST算法、BLAT算法等),将拼接后的序列比对至微生物参考基因组序列,从而获得与序列相关的物种分类信息。然而,不同于其他高等生物基因组的拼接,由于微生物的多样性,宏基因组研究中测序数据的拼接往往存在一定的难度,导致序列准确度不高。此外,传统的BLAST算法在对海量的拼接后数据进行比对时往往耗时较长,BLAST算法速率为目前二代测序常用的比对软件(如BWA、bowtie和SOAP等)速率的数百分之一至数十分之一,通常某些样本完成所有序列比对可能花费数天甚至更久的时间。因此笔者建议在实际分析过程中,使用二代测序的比对软件,把测序片段(不经过拼接)直接比对到参考物种的标识基因,从而快速获取物种分类信息及丰度信息,为后续的样本主坐标分析提供足够的信息。例如,目前国际人类微生物组计划项目中采用Metaphlan软件进行数据处理,该软件可以直接将测序序列比对至微生物参考数据库中(涵盖了美国生物技术信息中心基因组数据库中的2 887个微生物基因组数据),从而获得测序样本的菌群组成信息[21]。
3.8 不同分组样本的主坐标分析
主坐标分析是一种微生物组学数据分析中常用的降维及可视化方法,主要用于研究数据相似性或差异性,在微生物群落相关研究中,不同样本之间及不同环境之间微生物组成差异往往较大,所获得的物种丰度表通常由稀疏矩阵构成,因而不太适合使用其他组学数据通常采用的主成分分析方法。在主坐标分析中,首先对物种丰度组成的距离矩阵进行分解,获得一系列的特征值和特征向量,然后对特征向量进行排序,选择前几位的特征向量作为主要坐标,并将样品投影到这些向量的坐标轴上进行可视化展示。在对基于不同分类水平的物种丰度信息进行PCoA时,样本的物种组成越相似,它们在PCoA图上的距离越小。通过不同分组样本的主坐标分析,可以检测样本的测序质量以及深度是否提供了足够的信息来区别不同的分组样本,因而不同分组样本的主坐标分析是宏基因组数据分析质量控制流程中非常重要的步骤。
4 宏基因组数据质量控制应用实例
本文以一套已公开发表的肠道微生物宏基因组数据为例进行质量控制流程结果展示,该数据为正常人群与肝硬化人群肠道微生物队列研究数据,相应宏基因组测序数据下载自欧洲生物信息研究中心核酸数据库(ID:ERP005860)。本文选取部分有代表性的样本,对元数据和测序数据进行包括元数据整理、元数据和测序数据一致性检查、测序质量评估、低质量序列过滤、测序接头片段去除、宿主基因剔除、样本菌群组成分析、样本主坐标分析在内的质量控制检测。其原始数据测序片段的质量评估结果如图3所示,个别样本总体测序质量偏低,后续分析需加以控制或予以剔除。
质量控制前后样本原始数据及干净数据质量统计见表1(括号内为质量控制后的数值)。质量控制前后各样本重复序列百分比、序列平均长度及读段总数等均有明显变化,这表明了对原始测序数据进行相关质量控制的必要性。随后,笔者对各样本的菌群组成及微生物丰度进行评估,并在此基础上开展了样本的主坐标分析,结果如图4所示,正常样本与疾病样本可观察到一定的分布差异,表明了质量控制流程的有效性。

图3 测序片段不同位置碱基质量分布
5 宏基因组数据质量控制应用效果
上述宏基因组数据质量控制实例表明,本文提出的质量控制流程可以对宏基因组研究中的元数据和测序数据质量进行有效评估,从而为后续分析提供干净的数据。具体而言主要有以下几个方面。
● 元数据和测序数据的一致性检查保证了取样样本和测序样本的对应关系,并赋予测序样本表型信息,从而为测序样本的基因型和表型关联分析、不同分组样本的差异分析、样本菌群组成的机理研究分析提供支持。
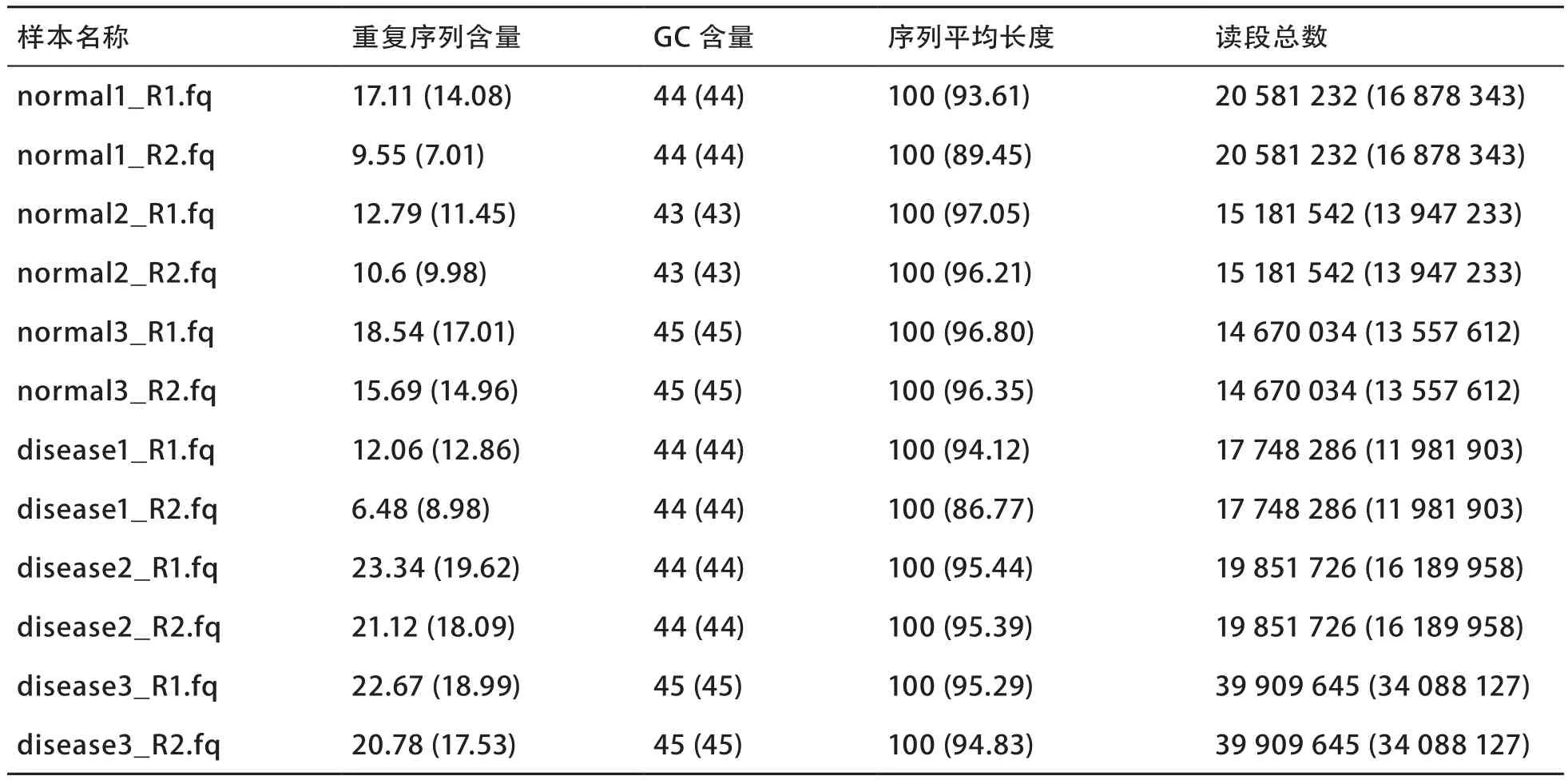
表1 质量控制前后序列质量统计
● 宏基因组数据质量控制流程中的质量检测、低质量测序序列的过滤及切除、接头序列及无关序列的剔除等步骤可以为后续的功能分析提供干净的分析数据,保证结果的可靠性。
● 宿主及污染序列的过滤,可以去除宿主基因片段对后续菌群组成分析的影响,保证分析的有效性。
● 不同分组样本的菌群组成和主坐标分析,可以在一定程度上评估样本的测序深度是否足够,即在当前的测序深度和质量条件下是否提供了足够的信息来满足分组样本差异分析的需求。
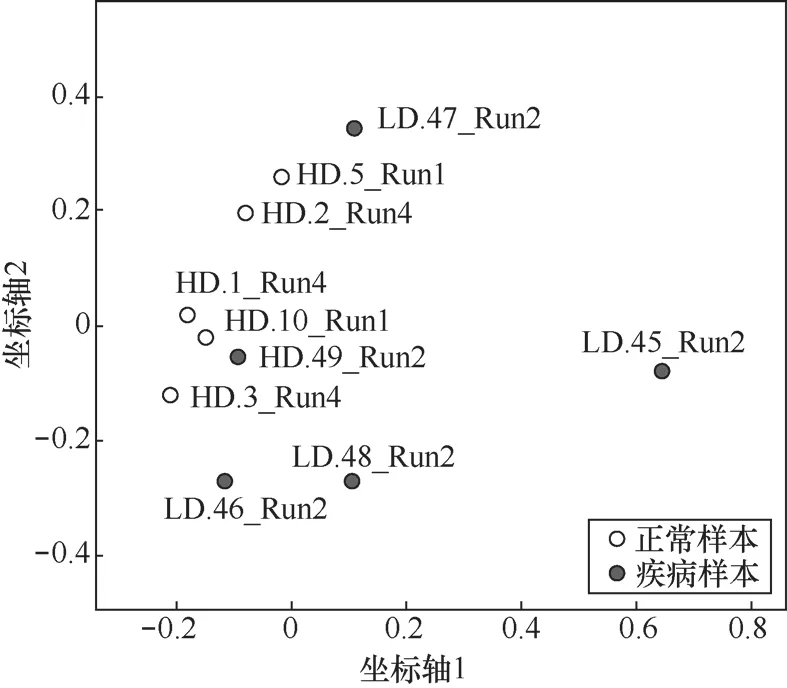
图4 主坐标分析结果
6 结束语
近年来,随着各种微生物组项目的开展,已有大量的宏基因组数据发布。如何充分利用和挖掘这些数据,对其进行更为深入的二次分析,从而获得新的发现,是一个极为重要的问题。宏基因组数据多分散在不同的数据库中或者不同的研究者手中,其数据收集和分析标准存在较大的差异,从而给数据的整合带来较大的障碍。如果能从生态类型(biotype)、数据类型(datatype)等角度系统整合这些数据,并提供统一的质量控制评价标准,将为宏基因组研究提供更大的便利。因此制定规范合理的数据质量控制标准,从而有效整合这些不同来源和类型的宏基因组数据,是未来的发展方向和趋势。本文对宏基因组数据分析过程中的质量控制流程进行了探讨,这将加速宏基因组学的相关研究。在此基础上,整合微生物的分类、进化、生态以及相关组学的数据,构建统一的微生物组数据仓库,并辅以微生物云服务平台,将解决微生物组研究各种数据分散在不同地方的局面,为微生物组大数据提供科学的管理机制和运行范式,从而为我国微生物组学研究提供技术支撑及基础大数据平台。
[1]张国庆, 宁康, 职晓阳, 等. 建设微生物组大数据中心发挥长期科学影响[J]. 中国科学院院刊, 2017(3): 280-289.ZHANG G Q, NING K, ZHI X Y, et al.Development of comprehensive microbiome big data warehouse/center for long-term scientific impact[J].Bulletin of Chinese Acadamy of Sciences, 2017(3): 280-289.
[2]HANDELSMAN J, RONDON M R, BRADY S F, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products[J]. Chemistry& Biology, 1998, 5(10): 245-249.
[3]WANG J, JIA H. Metagenome-wide association studies: fine-mining the microbiome[J]. Nat Rev Microbiol, 2016,14(8): 508-522.
[4]FANG H, CAI L, YU Y, et al. Metagenomic analysis reveals the prevalence of biodegradation genes for organic pollutants in activated sludge[J]. Bioresource Technology, 2013, 129(2): 209-218.
[5]DOS SANTOS H F, CURY J C, DO CARMO F L, et al. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution[J]. Plos One, 2011,6(3): e16943.
[6]QIN N, YANG F, LI A, et al. Alterations of the human gut microbiome in liver cirrhosis[J].Nature, 2014, 513(7516): 59-64.
[7]HE Z, PICENO Y, DENG Y, et al. The phylogenetic composition and structure of soil microbial communities shifts in response to elevated carbon dioxide[J].Isme Journal, 2012, 6(2): 259-272.
[8]CHEN Y E, TSAO H. The skin microbiome: current perspectives and future challenges[J]. Journal of the American Academy of Dermatology, 2013,69(1): 143-155.
[9]TURNBAUGH P J, LEY R E, HAMADY M,et al. The human microbiome project[J].Nature, 2007, 449(7164): 804-810.
[10]EHRLICH S D. Metagenomics of the intestinal microbiota: potential applications[J]. Gastroenterologie Clinique Et Biologique, 2010, 34(4S1):S23-S28.
[11]PAGANI I, LIOLIOS K, JANSSON J,et al. The Genomes OnLine Database(GOLD) v.4: status of genomic and metagenomic projects and their associated metadata[J]. Nucleic Acids Research, 2012(40): 571-579.
[12]FIELD D, GARRITY G, GRAY T, et al.The minimum information about a genome sequence (MIGS) specification[J]. Nature Biotechnology, 2008, 26(5): 541-547.
[13]KOTTMANN R, GRAY T, MURPHY,et al. A standard MIGS/MIMS compliant XML Schema: toward the development of the Genomic Contextual Data Markup Language (GCDML)[J]. Omics-a Journal of Integrative Biology, 2008, 12(2): 115-121.
[14]YILMAZ P, KOTTMANN R, FIELD D, et al.Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS)specifications[J]. Nature Biotechnology,2011, 29(5): 415-420.
[15]CARAUS I, ALSUWAILEM A A, NADON R, et al. Detecting and overcoming systematic bias in high-throughput screening technologies: a comprehensive review of practical issues and methodological solutions[J]. Briefings in Bioinformatics, 2015, 16(6): 97416.
[16]SCHMIEDER R, EDWARDS R. Quality control and preprocessing of metagenomic datasets[J]. Bioinformatics, 2011, 27(6):863-864.
[17]BOLGER A M, LOHSE M, USADEL B.Trimmomatic: a flexible trimmer for Illumina sequence data[J]. Bioinformatics,2014, 30(15): 2114-2120.
[18]SCHMIEDER R, YAN W L, ROHWER F,et al. TagCleaner: identification and removal of tag sequences from genomic and metagenomic datasets[J]. Bmc Bioinformatics, 2010, 11(1): 1-14.
[19]LI H, DURBIN R. Fast and accurate longread alignment with Burrows-Wheeler transform[J]. Bioinformatics, 2010, 26(5):589-595.
[20]LANGMEAD B, TRAPNELL C, POP M,et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome[J]. Genome Biology, 2009,10(3): R25.
[21]SEGATA N, WALDRON L, BALLARINI A,et al. Metagenomic microbial community profiling using unique clade-specific marker genes[J]. Nature Method, 2012, 9(8):811-814.
