叶绿体全基因组系统进化分析资源平台的构建
2018-06-01郑薇玮陈金慧施季森
郑薇玮,陈金慧,施季森
(1.福建江夏学院 电子信息科学学院,福建 福州 350108;2.南方现代林业协同创新中心,林木遗传与生物技术省部共建教育部重点实验室,南京林业大学 林学院,江苏 南京 210037)
1 引言
生物信息学是一门含括计算机科学、生命科学、数学、物理学、化学等在内的交叉学科领域[1].生物信息学研究利用数据库中海量的数据资源,辅以多种不同类型的数据处理工具来协同完成.生物信息学实验通常由一系列标准严格的步骤组成,激增的生物数据量使得实验过程变得更为复杂,单纯的手工实验难度加大[2].多数生物信息数据库在开发之初,就为研究人员开放了网络服务接口,允许不同的程序对数据资源进行直接访问.同时,生物信息学研究团队总是乐于无偿分享他们用于分析实验数据的程序和算法,网络上用于相同的目的的数据分析工具数量众多、良莠不齐.加强生物信息学在线资源(包括数据及其处理程序)的整合研究利用,搭建专用的生物信息研究平台,为生物学家的研究提供方便,被认为是生物信息技术发展的重要任务之一[3].
2 工作流技术
工作流是一种基于先进流程控制技术的信息系统.源自办公自动化的工作流系统,具有与办公自动化系统相独立的开发技术.办公自动化系统的核心目标是实现内部的流转,即能够使得办公过程中的业务环节实现相互衔接,上一个环节结束之后,处理结果能够自动流转到下一个环节[4].这种信息系统在实际应用中具有明显的优势,它支持应用逻辑和过程逻辑的分离[5],其中应用逻辑指的是管理模式中各环节的处理工作,过程逻辑指的是管理模式中各环节间的相互衔接.管理模式中的应用逻辑是基本不变的,而过程逻辑则根据具体的业务进行不断调整、变化的.通过对业务流程的有效整合和灵活设计,使信息系统能够适应大量不断变化的业务需求,在不修改具体功能应用的前提下,通过调整过程模式来实现系统概念下的功能改变[6].
工作流技术的研究工作已经在国内的许多领域开展,分布式应用技术(如J2EE、.NET等)的成熟为工作流的应用发展提供了更为有利的条件[7].新的工作流产品如雨后春笋般涌现,其应用主要关注业务流程管理和流程重组.由于工作流的基础定义至今未有定论,其标准规范也难以制定,目前存在众说纷纭、相互重叠的情况[7],导致人们常将工作流技术概念与其他技术相混淆,认为工作流只是其他某一概念的补充.
生物信息学领域,大量的数据、算法和程序工具散落分布在网络空间.工作流技术作为集成数据和算法的一个良好的解决方案,将其应用于生物信息学的研究分析,负责执行和监控生物信息学实验过程,能避免研究者们被复杂的编程环节困扰,从而将精力投放于了解生命现象本身.研究和开发基于不同生物信息学应用的工作流系统,具有重要的意义[8].
3 系统的实现
数据资源结构类型各异、地理位置分散,亟须一种能够快速获取、科学易用的工具对海量且繁杂的数据资源进行开发、共享和管理.资源目录的概念正是这样应运而生的.资源目录早期被应用在图书馆、档案、政务等信息资源的整合和组织中.例如,基于目录体系的政务信息资源,将分散的政务部门,按照领域、地区进行重新组织、按需整合,它们物理上分散、逻辑上集中,为使用者提供协同统一的政务信息,适应政务业务的动态变化,实现政务信息的整合共享.资源目录服务在业务需求驱动下,将网络环境中的相关资源按照目录树的结构进行整合和存储,网络资源即是它的目录信息,由它授权使用[9].本实验整合叶绿体基因组发育分析过程中所使用的在线平台,建立供实验室内部使用的叶绿体资源目录门户.
3.1 系统架构
叶绿体发育基因组学是植物进化生物学的发展趋势[10].叶绿体基因组与核基因组相比:结构保守;置换率适中;大小仅次于核基因组,且富集核苷酸与氨基酸序列信息;编码区与非编码区分子进化速率差异显著,适用于各阶层系统发育学研究.换言之,叶绿体作为系统发育学手段,具有明显的优势[10].同时,已有丰富的实验数据证明,叶绿体全基因组在植物系统发育关系的重建是行之有效的.例如,64种植物叶绿体基因组的联合进化分析,确认了Amborella为被子植物最基部类群[11].葡萄叶绿体全基因组与26种被子植物叶绿体基因组进行进化分析,显示葡萄科与蔷薇分支有姐妹亲缘[12].
生物信息学实验涉及大量的严格统一的工作步骤,需要多种不同类型的数据和工具协同完成.整合和管理分布广泛的程序、算法和Web服务,高效完成生物信息学分析具有重要意义.已有大量的面向实际问题的生物信息学工作流,如序列分析、基因芯片数据差异表达分析等科学计算系统被成功构建,基于叶绿体全基因组的进化分析系统则鲜有报道.本实验是在实验室进行杉木系统发育地位确认的相关实验过程中,收集了基于叶绿体基因组构建系统发育图谱流程的相关在线服务,并进行归类整合,构建的在线资源门户,系统结构图如图1所示.根据实际工作需要,设计基于工作流的进化分析资源目录采用三层的B/S模式架构,在逻辑上分为表示层、应用层、数据层三层,如图2所示.
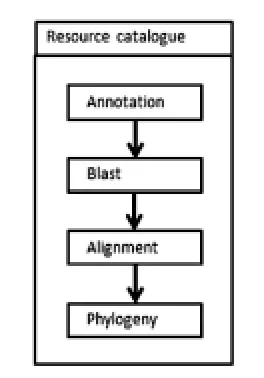
图1 总体结构图
3.2 系统功能模块设计
如图1所示,根据叶绿体基因组进化发育分析流程的4个步骤,从在线服务中将如下四个模块进行整合,并使用jQuery技术来实现其菜单导航,即:对测序后的叶绿体基因组使用DOGMA(http://dogma.ccbb.utexas.edu)进行注释;对未被注释的基因进行Blast(http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=tblastn&PAGE_TYPE=BlastSearch&BLAST_SPEC=&LINK_LOC=blasttab&LAST_PAGE=blastx)比对,寻找数据库中的已注释过的同源基因;Alignment(http://www.ebi.ac.uk/Tools/msa/muscle/),从而找出最佳核苷酸迭代模型;将模型导入工具,进行系统进化分析Phylogeny(http://www.phylogeny.fr/one_task.cgi?task_type=mrbayes).
3.3 工作流设计
XML作为一种数据描述语言,允许用户根据需要自定义标记及属性.在我们的资源目录中采用XML来描述系统工作流程配置.如图1所示的,叶绿体基因组的进化分析4个过程任务,分别对应系统的4个功能模块.使用XML描述的工作流程如图3所示.其中使用<Cworkflow></Cworkflow>标识一个工作流定义块;<Cprocess></Cprocess>定义需要执行的业务流程;<Csection></Csection>用于标识系统功能模块.

图3 工作流程
3.4 系统部署
为了验证这种基于工作流技术的生物信息系统的可靠性、稳定性,将系统部署到实验室GNU/LINUX操作系统大型服务器上,软件环境使用jdk1.8.0_74+apache-tomcat-8.0.23容器作为测试环境.客户端只需要安装有浏览器的普通PC机即可.
4 结语
由于生物信息分析过程其实质也是模块化、流程化的数据处理过程,工作流技术被认为在生物信息学研究中有很大的发展空间.目前,大量面向实际生物信息应用问题的工作流,如基因差异表达分析、序列表达等已经被构建[13].这些工作流管理系统数量多、标准不一,各个系统定义工作流的语言不同、难于统一.
本实验尝试构建基于当前叶绿体基因组序列分析工具的资源目录工作流系统,将各分析功能模块有机整合,系统灵活易用、集成成本低、易扩展,为本实验室的科研人员在叶绿体基因组研究方面提供支持.由于,本平台仅供实验室内部使用,在构建系统的时候,尚未考虑网络环境的复杂性,数据源也是有所选择和针对性(即其结构和数量均稳定).在今后的工作中,将在上述几个方面做进一步考虑.
〔1〕 李淮.生物信息学研究概述[J].开封教育学院学报,2017,37(6):283-284.
〔2〕 Li P,Oinn T,Soiland S,et al.Automated manipulation of systems biology models using libSBML within Taverna workflows[J].Bioinformatics,2007,24(2):287-289.
〔3〕 吴占坤.电子政务中的信息资源整合研究[D].哈尔滨理工大学,2007.
〔4〕 杜鹏.工作流技术及其在办公自动化系统中的应用.万方数据资源系统,2008.
〔5〕 谭宁.基于Petri网的办公自动化系统结构模型设计[J].硅谷,2009(12):53-53.
〔6〕 顾煜新,张淑华,方艳,等.基于工作流技术的网络平行审计系统的研究[J].长春大学学报,2006,16(8):56-59.
〔7〕 黄钰.工作流BioW平台的构建与应用[D].华中科技大学,2008.
〔8〕 Romano P.Automation of in-silico data analysis processes through workflow management systems[J].Briefings in Bioinformatics,2007,9(1):57-68.
〔9〕 肖侬,任浩,徐志伟,等.基于资源目录技术的网格系统软件设计与实现[J].计算机研究与发展,2002,39(8):902-906.
〔10〕 张韵洁,李德铢.叶绿体系统发育基因组学的研究进展[J].植物分类与资源学报,2011,33(4):365-375.
〔11〕 Jansen RK,Cai Z,Raubeson LA,et al.Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns[J].Proc Natl Acad Sci U S A,2007,104(49):19369-19374.
〔12〕 Jansen RK,Kaittanis C,Saski C,et al.Phylogenetic analyses of Vitis (Vitaceae)based on complete chloroplast genome sequences:effects of taxon sampling and phylogenetic methods on resolving relationships among rosids[J].Bmc Evolutionary Biology,2006,6(1):32.
〔13〕 闵波,张乐平,刘小宇,等.面向生物信息学的工作流管理系统框架[J].生物信息学,2011(3):213-216.
