邻亚甲基醌和3-氯吲哚啉酮[4+1]环加成合成螺环吲哚啉酮
2018-05-30杜升华周吉江国防
杜升华,周吉,江国防
邻亚甲基醌和3-氯吲哚啉酮[4+1]环加成合成螺环吲哚啉酮
杜升华1, 2,周吉2,江国防2
(1. 湖南化工研究院有限公司,湖南 长沙,410014;2. 湖南大学 化学化工学院,湖南 长沙,410082)
以3-氯吲哚啉酮与邻亚甲基醌中间体为原料,在碱性条件下通过Michael加成/环化串联反应,高收率、高非对映选择性地构建双杂环螺环化合物。其中,邻亚甲基醌中间体通过磺酰基取代苯酚在无机碱作用下原位生成获得,在温和、简易操作条件下与3-氯吲哚啉酮作用一步制备含有苯并呋喃和吲哚啉酮这2种重要杂环骨架结构的螺环化合物。通过条件筛选,在最优条件下获得目标产物。为了验证此方法的实用性,进行克级规模试验。研究结果表明:在最优条件下,目标产物收率高达92%,非对映选择性r大于20:1;扩大底物用量,收率和非对映选择性仍很高;此方法普适性广,对于多种类型的磺酰基取代苯酚以及吲哚啉酮底物同样适用。
3-氯吲哚啉酮;邻亚甲基醌中间体;Michael加成/环化串联反应;螺环吲哚啉酮化合物
含有吲哚啉酮模块的螺环产物由于其在天然产物和化学中间体合成中的重要作用,近年来受到了人们的青睐[1−3]。许多生物活性分子含有这种骨架结构并显示出多种多样的生物活性,如图1中化合物A具有良好的抗细菌活性[4],化合物B是一类重要的非核苷类逆转录酶抑制剂[5],而化合物C则具有抗肿瘤活性[6]。基于含有吲哚啉酮模块的螺环产物的重要性,迫切需要发展高效、温和的方法以构建这种骨架结构。3-氯吲哚啉酮由于其在特定条件下容易异构从而同时具有亲电与亲核性,因此,其被广泛应用于各类取代以及加成反应中以构建具有吲哚啉酮骨架结构的衍生物[7−9]。近年来,LI等[10]运用3-氯吲哚啉酮构建螺环吲哚啉酮并取得了一系列成果。邻亚甲基醌中间体是一类化学性质极其活泼的去芳香性反应中间体,被广泛应用于有机合成、药物化学和材料化学中。该中间体由于其结构特殊,存在电荷分离的极限共振式,在热力学驱动力下,利于恢复芳香性而具有较高的反应活性,能够与各种取代的烯烃发生分子间Diels-Alder反应、与各种亲核试剂发生亲核加成和环化反应[11−16]以及自身聚合反应,生成含各种取代基的杂环化合物,这些化合物在天然产物的合成中具有重要作用[17−19]。CAO 等[20−26]对吲哚相关杂环化合物的合成进行了研究,本文作者在此基础上运用碱性条件下原位生成的邻亚甲基醌中间体与3-氯吲哚啉酮发生Michael加成/环化串联的[4+1]环加成反应,以高收率高非对映选择性地构建同时含有苯并呋喃与吲哚啉酮骨架结构的双杂环螺环产物。

图1 具有生物活性的螺环吲哚啉酮分子
1 实验
1.1 实验原理
运用原位生成的邻亚甲基醌中间体与3-氯吲哚啉酮反应构建螺环吲哚啉酮产物的原理如图2所示。磺酰基取代苯酚在无机碱作用下原位生成邻亚甲基醌中间体,其与3-氯吲哚啉酮底物发生Michael加成/环化串联的[4+1]环加成反应,从而高收率、高非对映选择性地构建含有双杂环的螺环产物。
1.2 实验方法
以5-甲氧基-2-[苯基(磺酰基)甲基]-苯酚(1a)与3-氯吲哚啉酮(2a)为模型底物进行反应,反应方程式为

实验结果如表1所示。在空气中,向反应瓶中依次加入磺酰烷基取代苯酚1,3-氯吲哚啉酮2,碳酸钠以及1,2-二氯乙烷(DCE),置于温度为50 ℃的水浴锅中反应36~72 h,利用薄层色谱分析法检测反应完全之后进行柱层析分离。在所用淋洗液中,乙酸乙酯与石油醚体积比为2:1。利用旋转蒸发仪除去溶剂,将油泵抽干后即得目标螺环产物6-甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3′-吲哚啉]-2′-酮(3aa)。
1.3 分析仪器
分析仪器为:BRUKER DRX 400核磁仪;Applied Biosystems Mariner System 5303 HRMS。
2 结果与讨论
2.1 反应条件优化
首先,以碳酸钾为碱,在二氯甲烷中,于室温下反应36 h,以39%的分离收率以及非对映异构体比例r1:1获得目标产物螺环吲哚啉酮3a。当选用碱性较强的碳酸铯以及氢氧化钠时,体系较杂且非对映选择性差,但使用碳酸钠时非对映异构体比例r能达到12:1,因此,选取碳酸钠为最佳碱。当温度升高至40 ℃时,产物非对映选择性r大于20:1。随后,在40 ℃筛选一系列常用有机溶剂,在甲苯、乙腈以及氯代溶剂中,非对映选择性r都大于20:1,但甲苯和乙腈中反应活性不太理想,因此,选择沸点较高的1,2二氯乙烷作为最佳溶剂进一步筛选。继续提高温度,发现当温度高于50 ℃时,非对映选择性r变小而且反应体系较杂,导致目标产物分离收率低,在50 ℃能得到最高的分离收率95%以及非对映异构体比例r大于20:1。因此,最优条件为:以DCE为溶剂在50 ℃反应36 h。
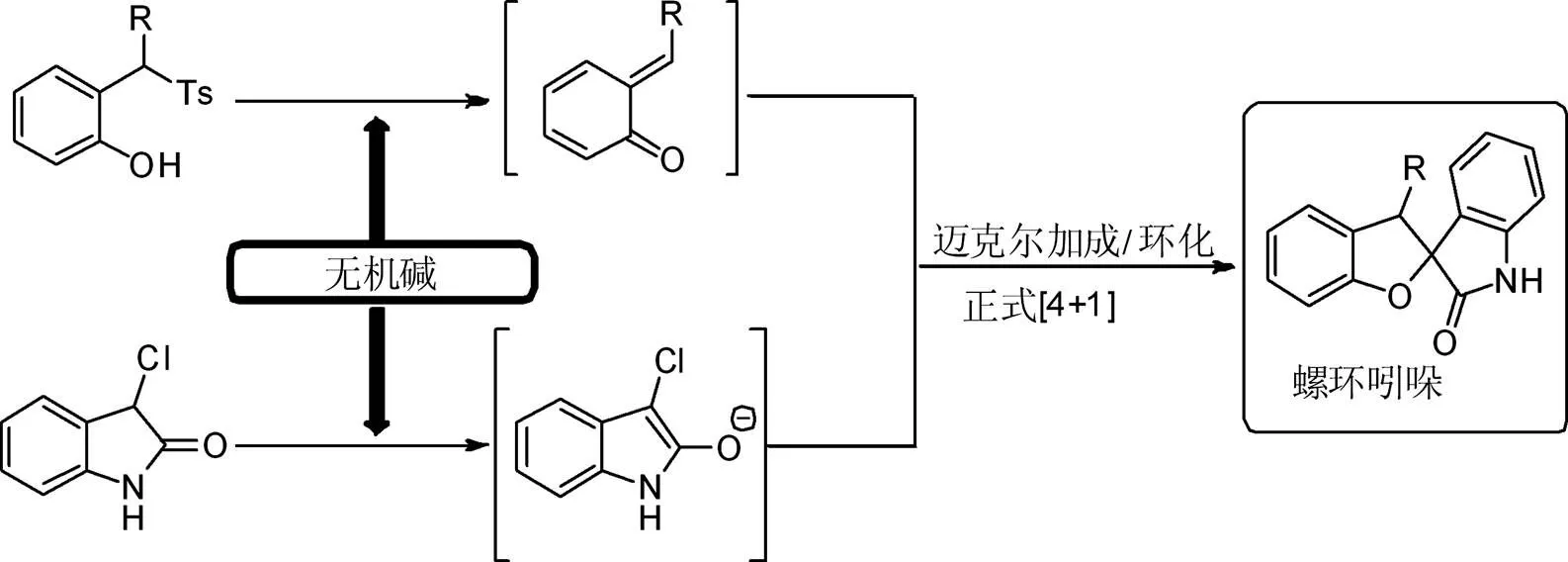
图2 运用原位生成的邻亚甲基醌中间体与3-氯吲哚啉酮反应构建螺环吲哚啉酮产物的原理
Fig. 2The principle of usinggenerated-quinone methides intermediates to react with 3-chloroindolinone to construct spirocyclic porphyrinone products
2.2 磺酰基取代苯酚底物拓展
在确定了最佳条件后,对反应底物中的磺酰基取代苯酚底物1的范围进行拓展,如图3所示。首先,对磺酰基取代苯酚苄位上的不同取代芳基进行研究,发现不论是电子效应还是位阻效应对反应的影响都很小,最终都能高产率的得到对应的目标产物3aa,3ba,3ca,3da,3ea,3fa和3ga。随后,改变苯酚骨架结构分别为萘酚底物1h以及双甲氧基取代底物1i,反应能正常进行,并以高收率和高非对映选择性获得对应产物3ha和3ia。但在苯酚苯环上引入吸电子基团后时反应基本不能发生,其原因可能是当苯环上引入吸电子基团后不利于邻亚甲基醌中间体生成,导致无法获得目标产物。
2.3 3-氯吲哚啉酮底物拓展
为了进一步考察底物的普适性,对反应底物中的3-氯吲哚啉酮底物2的底物范围进行拓展,如图4所示。首先,在吲哚5位分别引入给电子基团甲基和吸电子基团溴和氯,反应都能顺利进行并获得较高收率和高非对映选择性的目标产物(3ab,3ac和3ad)。同样,在吲哚4位以及6位引入吸电子的卤素原子,也能获得目标产物(3ae,3af,3ag和3ah)。最后,同时变化磺酰基取代苯酚底物和3-氯吲哚啉酮底物取代基,不同底物也可以成功组合反应,以中等到较高的收率获得螺环吲哚啉酮3bf和3ih。需指出的是:在这些底物中,卤素的引入为进一步化学转化提供了可能的反应位点。但由于化合物2本身性质较活泼,导致其他强吸电子基团和给电子基团取代的底物和7位取代的底物目前无法合成。
2.4 克级规模试验
当使用模型底物1a(1.326 g, 3.6 mmol)和2a (0.503 g, 3.0 mmol)反应时,同样能以92%的收率和大于20:1的非对映选择性获得对应螺环吲哚啉酮产物3aa (0.947 g)。
2.5 产物3aa相对构型的确定
将10 mg产物3aa溶于0.5 mL二氯甲烷和0.5 mL甲苯混合溶剂中,向其中缓慢加入5 mL正己烷,完毕后将体系静置于阴凉处,4 d后析出透明针状固体。通过单晶衍射分析确定3aa相对构型如图6所示,其对应的CCDC编号为1487826,具体晶体参数可通过网址www.ccdc.com.ac.uk/data_request/cif免费获得。

表1 条件优化
a. 反应条件:磺酰烷基取代苯酚1a(0.24 mmol),3-氯吲哚啉酮2a(0.20 mmol),碱(0.48 mmol),溶剂(3.0 mL),36 h;b.据1H NMR分析确定;c. 分离后的产率;d. 反应时间为12 h。

图3 磺酰基取代苯酚底物拓展
2.6 反应机理
以模型底物反应为例,根据实验结果得出可能的反应机理为:首先,在无机碱作用下,吲哚啉酮2a异构为烯醇式对原位生成的邻亚甲基醌中间体进行共轭加成得到中间体I,紧接着发生亲核取代反应实现关环,得到目标产物螺环吲哚啉酮3aa。净结果为[4+1]环加成反应。
2.7 产物数据表征
1) 6-甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3aa):65 mg,收率为95%,浅黄色固体,熔点为108~109oC,比移值f(即组分的迁移距离与展开剂的迁移距离之比)为0.60 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.68(单峰,1H),7.54(双峰,耦合常数=7.4 Hz,1H),7.29(td峰,耦合常数分别为7.7 Hz和1.1 Hz,1H),7.22~7.10(多重峰,4H),7.00(td峰,耦合常数分别为7.9 Hz和1.1 Hz,3H),6.69(双峰,耦合常数=7.8 Hz,1H),6.59(双峰,耦合常数=2.2 Hz,1H),6.55(dd峰,耦合常数=8.2,2.3 Hz,1H),5.09(单峰,1H),3.82(单峰,3H)。
13C NMR(100 MHz,CDCl3):化学位移分别为175.0,161.5,161.2,141.4,135.2,130.6,128.9,128.6,128.3,128.0,125.2,124.4,123.2,119.9,110.4,107.3,96.8,91.8,58.5和55.6。
对高分辨质谱进行计算,得出此分子C22H18NO3[M+H]+质谱相对分子质量为344.120 8,在相对分子质量为344.120 7处找到实际峰。

图4 3-氯吲哚啉酮底物的拓展

图5 克级规模试验
2) 6-甲氧基-3-(4-甲基苯基)-3H-螺环[苯并呋 喃-2,3'-吲哚啉]-2'-酮(3ba):70 mg,收率为97%,白色固体,熔点为98~99℃,f为0.65 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.64(单峰,1H),7.53(双峰,耦合常数=7.4 Hz,1H),7.29(三重峰,耦合常数=7.7 Hz,1H),7.13(三重峰,耦合常数=7.5 Hz,1H),7.04~6.93(多重峰,3H),6.90(双峰,耦合常数=8.0 Hz,2H),6.69(双峰,耦合常数=7.8 Hz,1H),6.63~6.47(多重峰,2H),5.06(单峰,1H),3.82(单峰,3H),2.24(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.9,161.5,161.1,141.3,137.6,132.0,130.6,129.0,128.8,128.7,125.2,124.5,123.2,120.1,110.3,107.2,96.8,91.8,58.2,55.6和21.1。
对高分辨质谱进行计算,得出此分子C23H20NO3[M+H]+质谱相对分子质量为358.136 5,在相对分子质量为358.136 2处找到实际峰。

图6 产物3aa相对构型的确定

图7 反应机理
3) 6-甲氧基-3-(3-甲基苯基)-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ca):70 mg,收率为97%,无色油状液体,f为0.65(正己烷与乙酸乙酯体积比为 2:1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.60(单峰,1H),7.53(双峰,耦合常数= 7.4 Hz,1H),7.29(三重峰,耦合常数=7.7 Hz,1H),7.14(三重峰,耦合常数=7.5 Hz,1H),7.08~6.96(多重峰,3H),6.85(单峰,1H),6.78(双峰,=7.1 Hz,1H),6.70(双峰,=7.8 Hz,1H),6.67~6.50(多重峰,2H),5.06(单峰,1H),3.82(单峰,3H),2.19(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.7,161.5,161.1,141.3,137.9,135.1,130.6,129.5,128.7,128.7,128.1,126.0,125.3,124.5,123.2,120.0,110.2,107.3,96.8,91.8,58.5,55.6和21.4。
对高分辨质谱进行计算,得出此分子C23H20NO3[M+H]+质谱相对分子质量为358.136 5,在相对分子质量为358.136 6处找到实际峰。
4) 6-甲氧基-3-(4-甲氧基苯基)-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3da):70 mg,收率为94%,橙色油状液体,f为0.45(正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.73(单峰,1H),7.53(双峰,耦合常数=7.4 Hz,1H),7.29(三重峰,=7.7 Hz,1H),7.13(三重峰,=7.5 Hz,1H),6.95(dd峰,=15.3,8.4 Hz,3H),6.74~6.62(多重峰,3H),6.62-6.48(多重峰,2H),5.04(单峰,1H),3.82(单峰,3H),3.69(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为 175.0,161.4,161.1,159.2,141.3,130.6,130.0,128.6,126.9,125.1,124.5,123.2,120.3,113.7,110.3,107.2,96.8,91.8,57.9,55.6和55.1。
对高分辨质谱进行计算,得出此分子C23H20NO4[M+H]+质谱相对分子质量为374.1314,在相对分子质量为374.131 2处找到实际峰。
5) 6-甲氧基-3-(3-甲氧基苯基)-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ea):72 mg,收率为96%,无色油状液体,f为0.55 (正己烷与乙酸乙酯体积比为 2:1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.78(单峰,1H),7.55 (双峰,耦合常数=7.5 Hz,1H),7.33(三重峰,耦合常数=7.6 Hz,1H),7.11(三重峰,耦合常数=7.6 Hz,1H),6.85(dd峰,耦合常数分别为=15.3,8.6 Hz,3H),6.72~6.60(多重峰,3H),6.62~6.48(多重峰,2H),5.08(单峰,1H),3.85(单峰,3H),3.73(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为175.2,161.3,161.2,159.1,141.5,130.5,130.2,128.4,126.7,125.0,124.4,123.1,120.2,113.4,110.1,107.1,96.6,91.7,57.9,55.6和55.2。
对高分辨质谱进行计算,得出此分子C23H20NO4[M+H]+质谱相对分子质量为374.131 4,在相对分子质量为374.131 8处找到实际峰。
6) 6-甲氧基-3-(4-三氟甲基苯基)-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3fa):73 mg,收率为90%,白色固体,熔点为65~66℃,f为0.65 (正己烷与乙酸乙酯体积比为2/1)。1H NMR (400 MHz,CDCl3):化学位移分别为7.54 (单峰,1H),7.50~7.39(多重峰,3H),7.33(三重峰,耦合常数=7.7 Hz,1H),7.16(三重峰,耦合常数=7.7 Hz,3H),6.96(双峰,耦合常数= 8.1 Hz,1H),6.72(双峰,耦合常数=7.8 Hz,1H),6.64-6.52(多重峰,2H),5.14(单峰,1H),3.83(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.2,161.5,161.5,141.0,139.6,130.9,130.3,130.0,129.4,128.2,125.2(四重峰,耦合常数= 3.7 Hz),125.1,124.5,123.5,119.1,110.4,107.6,96.9,91.4,58.2和55.6。
19F NMR (376 MHz,CDCl3)化学位移为−62.57。
对高分辨质谱进行计算,得出此分子C23H17F3NO3[M+H]+质谱相对分子质量为412.1082,在相对分子质量为412.108 1处找到实际峰。
7) 3-(4-氟苯基)-6-甲氧基-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3ga):71 mg,收率为97%,白色固体,熔点120~121℃,f为0.70 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.76(单峰,1H),7.53(双峰,耦合常数=7.4 Hz,1H),7.30(三重峰,耦合常数=7.7 Hz,1H),7.14(三重峰,=7.6 Hz,1H),7.04~6.92(多重峰,3H),6.85(三重峰,耦合常数=8.6 Hz,2H),6.71 (双峰,耦合常数= 7.8 Hz,1H),6.63~6.50(多重峰,2H),5.06(单峰,1H),3.82(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.8,162.5(双峰,耦合常数C-F=245.0 Hz),161.4,161.3,141.2,130.9(双峰,耦合常数C-F=3.2 Hz),130.8,130.6(双峰,耦合常数C-F=8.1 Hz),128.4,125.1,124.5,123.4,119.8,115.2(双峰,耦合常数C-F= 21.2 Hz),110.3,107.4,96.8,91.7,57.8和55.6。
19F NMR (376 MHz,CDCl3):化学位移为−114.14。
对高分辨质谱进行计算,得出此分子C22H17FNO3[M+H]+质谱相对分子质量为362.111 4,在相对分子质量为362.111 3处找到实际峰。
8) 3'-苯基-3'H-螺环[吲哚啉-3,2'-萘酚[1,2-b]呋喃]-2-酮(3ha):68 mg,收率为94%,黄色固体,熔点为165~166oC,f为0.75 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为8.01 (td峰,耦合常数分别为=7.1,3.6 Hz,1H),7.92(单峰,1H),7.90~7.83(多重峰,1H),7.55~7.44(多重峰,4H),7.25~7.13(多重峰,6H),7.10(dd峰,耦合常数分别为9.5 Hz和5.5 Hz,1H),7.04(dd峰,耦合常数分别为7.7 Hz和1.5 Hz,2H),6.64(三重峰,耦合常数= 7.3 Hz,1H),5.34(单峰,1H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.7,155.9,141.3,135.6,134.7,130.6,129.4,129.1,128.4,128.0,128.0,126.4,125.8,124.4,123.3,122.5,121.8,121.5,121.0,120.7,110.5,91.6和60.0。
对高分辨质谱进行计算,得出此分子C25H18NO2[M+H]+质谱相对分子质量为364.125 9,在相对分子质量为364.125 8处找到实际峰。
9) 5,6-双甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3ia):69 mg,收率为92%,黄色固体,熔点为134~135 ℃,f为0.45 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.72 (单峰,1H),7.55 (双峰,耦合常数=7.4 Hz,1H),7.31(三重峰,耦合常数=7.7 Hz,1H),7.24~7.11(多重峰,4H),7.04(双峰,耦合常数=6.7 Hz,2H),6.69(dd峰,耦合常数分别为17.3 Hz和7.4 Hz,3H),5.14(单峰,1H),3.90(单峰,3H),3.80(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为176.8,156.3,152.1, 146.2,143.1,137.2,132.5,130.8,130.7,130.3,129.9,126.3,125.1,119.6,112.2,110.8,97.3,93.4,61.4,58.8和58.0。
对高分辨质谱进行计算,得出此分子C23H20NO4[M+H]+质谱相对分子质量为374.131 4,在相对分子质量为374.131 4处找到实际峰。
10) 6-甲氧基-5'-甲基-3-苯基-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ab):65 mg,收率为90%,无色油状液体,f为0.55 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.70(单峰,1H),7.43(双峰,耦合常数=7.4 Hz,1H),7.18(三重峰,耦合常数=7.7 Hz,1H),7.10(三重峰,耦合常数=7.5 Hz,1H),7.09~6.92(多重峰,3H),6.88(单峰,1H),6.78(双峰,耦合常数=7.1 Hz,1H),6.68(双峰,耦合常数=7.8 Hz,1H),6.59~6.48(多重峰,2H),5.06(单峰,1H),3.82(单峰,3H),2.26(单峰,3H)。
13C NMR (100 MHz,CDCl3) 化学位移分别为172.8,161.6,161.8,143.9,135.8,130.4,130.0,129.7,128.7,126.5,125.4,124.5,123.1,119.8,110.0,107.3,96.9,91.7,58.8,55.5和21.0。
对高分辨质谱进行计算,得出此分子C23H20NO3[M+H]+质谱相对分子质量为358.136 5,在相对分子质量为358.136 6处找到实际峰。
11) 5'-溴-6-甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3ac):80 mg,收率为95%,红色油状液体,f为0.60 (正己烷与乙酸乙酯体积比为 2/1)。1H NMR (400 MHz,CDCl3):化学位移分别为7.26(单峰,1H),7.30(双峰,耦合常数=8.5 Hz,1H),7.24~7.12(多重峰,4H),7.05(dt峰,耦合常数分别为13.3 Hz和10.6 Hz,3H),6.69(双峰,耦合常数=7.8 Hz,1H),6.59(双峰,耦合常数= 2.0 Hz,1H),6.56(dd峰,耦合常数分别为8.5 Hz和2.0 Hz,1H),5.58(单峰,1H),3.80(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.2,162.7,162.1,142.9,136.4,132.8,131.9,129.1,128.0,128.0,127.5,125.0,120.1,119.5,108.9,108.0,96.6,92.1,55.5和54.6。
对高分辨质谱进行计算,得出此分子C22H17BrNO3[M+H]+质谱相对分子质量为422.031 4,在相对分子质量为422.031 5处找到实际峰。
12) 5'-氯-6-甲氧基-3-苯基-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ad):71 mg,收率为94%,白色固体,熔点为133~134℃,f为0.50 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为8.06(单峰,1H),7.51(双峰,耦合常数=1.9 Hz,1H),7.25(双峰,耦合常数=2.0 Hz,1H),7.21~7.11(多重峰,3H),7.04~6.92(多重峰,3H),6.65~6.49(多重峰,3H),5.05(单峰,1H),3.82(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.8,161.3,139.8,134.9,130.6,130.4,128.9,128.4,128.4,128.1,125.3,124.8,119.5,111.6,107.5,96.8,91.6,58.6和55.6。
对高分辨质谱进行计算,得出此分子C22H17ClNO3[M+H]+质谱相对分子质量为378.081 9,在相对分子质量为378.082 1处找到实际峰。
13) 4'-氟-6-甲氧基-3-苯基-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ae):68 mg,收率为94%,黄色固体,熔点为77~78℃,f为0.65 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.65(单峰,1H),7.29(td峰,耦合常数分别为 8.2 Hz和5.5 Hz,1H),7.23~7.14(多重峰,3H),7.10~7.04(多重峰,2H),7.00(双峰,耦合常数=8.2 Hz,1H),6.83(三重峰,耦合常数=8.8 Hz,1H),6.59(三重峰,耦合常数=3.9 Hz,1H),6.58~6.52(多重峰, 1H),6.50(双峰,耦合常数=7.8 Hz,1H),5.36 (双峰,耦合常数=12.9 Hz,1H),3.82(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为174.0,161.4,159.8(双峰,耦合常数C-F=252.7 Hz),143.1(双峰,耦合常数C-F=7.9 Hz),135.1,132.8(双峰,耦合常数C-F=3.8 Hz),129.1,128.4,128.0,125.3,119.7,114.1,114.0,110.8(双峰,耦合常数C-F=19.9 Hz),107.4,106.5,106.5,96.8,91.0和55.6。
19F NMR (376 MHz,CDCl3):化学位移为−116.15。
对高分辨质谱进行计算,得出此分子C22H17FNO3[M+H]+质谱相对分子质量为362.111 4,在相对分子质量为362.111 5处找到实际峰。
14) 4'-溴-6-甲氧基-3-苯基-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3af):80 mg,收率为95%,橙色油状液体,f为0.60 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.36(单峰,1H),7.30~7.26(多重峰,1H),7.18(tt峰,耦合常数分别为8.0 Hz和4.0 Hz,4H),7.08~6.98(多重峰,3H),6.67(双峰,耦合常数=7.7 Hz,1H),6.62(双峰,耦合常数=2.1 Hz,1H),6.56(dd峰,耦合常数分别为8.3 Hz和2.2 Hz,1H),5.60(单峰,1H),3.83(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为173.8,161.7,161.1,143.0, 135.4,133.1,131.9,129.1,128.3,128.0,127.2,125.2,120.1,119.5,109.3,107.3,96.6,92.3,55.6和54.6。
对高分辨质谱进行计算,得出此分子C22H17BrNO3[M+H]+质谱相对分子质量为422.031 4,在相对分子质量为422.031 7处找到实际峰。
15) 6'-溴-6-甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3'吲哚啉]-2'-酮(3ag):78 mg,收率为93%,浅红色固体,熔点为105~106 ℃,f为0.85 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.88(单峰,1H),7.40(双峰,耦合常数=8.0 Hz,1H),7.32~7.26(多重峰,1H),7.23~7.12(多重峰,3H),7.00(三重峰,耦合常数=7.2 Hz,3H),6.87(双峰,耦合常数=1.3 Hz,1H),6.62~6.50(多重峰,2H),5.05(单峰,1H),3.82(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为176.6,163.2,163.2,144.4,136.8,130.8,130.3,130.0,129.5,128.2,127.7,127.2,126.2,121.4,115.8,109.4,98.7,93.2,60.4和57.5。
对高分辨质谱进行计算,得出此分子C22H17BrNO3[M+H]+质谱相对分子质量为422.031 4,在相对分子质量为422.031 6处找到实际峰。
16) 6'-氯-6-甲氧基-3-苯基-3H-螺环[苯并呋喃- 2,3'-吲哚啉]-2'-酮(3ah):68 mg,收率为90%,淡黄色固体,熔点为142~143℃,f为0.80 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.67(单峰,1H),7.47(双峰,耦合常数=8.0 Hz,1H),7.16(ddd峰,耦合常数分别为11.5,9.3和4.4 Hz,4H),7.07~6.92(多重峰,3H),6.73(单峰,1H),6.63~6.48(多重峰,2H),5.06(单峰,1H),3.82(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为176.5,163.2,163.2,144.2,138.3,136.8,130.8,130.3,130.0,128.9,127.5,127.1,125.2,121.4,112.9,109.4,98.7,93.2,60.5和57.5。
对高分辨质谱进行计算,得出此分子C22H17ClNO3[M+H]+质谱相对分子质量为378.081 9,在相对分子质量为378.082 0处找到实际峰。
17) 4'-溴-6-甲氧基-3-(4-甲基苯基)-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3bf):80 mg,收率为92%,紫色油状液体,f为0.35 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.70(单峰,1H),7.50(双峰,耦合常数=7.2 Hz,1H),7.29(三重峰,耦合常数=7.7 Hz,1H),7.22(三重峰,耦合常数=7.5 Hz,1H),7.02~6.98(多重峰,3H),6.90(双峰,耦合常数=7.8 Hz,2H),6.60~6.48(多重峰,2H),5.11(单峰,1H),3.79(单峰, 3H),2.19(单峰,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为175.1,162.2,161.0,141.2,137.9,132.5,130.5,129.8,128.7,128.9,125.2,124.6,123.0,120.1,110.5,107.5,96.7,91.8,58.2,55.9和21.1。
对高分辨质谱进行计算,得出此分子C23H19BrNO3[M+H]+质谱相对分子质量为436.047 0,在相对分子质量为436.047 1处找到实际峰。
18) 6'-氯-5,6-双甲氧基-3-苯基-3H-螺环[苯并呋喃-2,3'-吲哚啉]-2'-酮(3ih):70 mg,收率为86%,黄色固体,熔点为88~89℃,f为0.50 (正己烷与乙酸乙酯体积比为 2/1)。
1H NMR (400 MHz,CDCl3):化学位移分别为7.68(单峰,1H),7.47(双峰,耦合常数=8.0 Hz,1H),7.25~7.18(多重峰,3H),7.13(dd峰,耦合常数分别为8.0 Hz和1.8 Hz,1H),7.05~6.98(多重峰,2H),6.73(dd峰,耦合常数分别为=7.3,2.1 Hz,1H),6.64(三重峰,耦合常数=6.1 Hz,2H),5.09(单峰,1H),3.89(单峰,3H),3.79(s,3H)。
13C NMR (100 MHz,CDCl3):化学位移分别为176.6,156.1,152.2,146.4,144.2,138.2,136.9,130.8,130.4,130.1,129.1,127.4,125.2,119.2,112.9,110.7,97.3,92.9,61.4,58.8和58.1。
对高分辨质谱进行计算,得出此分子C23H19ClNO4[M+H]+质谱相对分子质量为408.092 4,在相对分子质量为408.092 5处找到实际峰。
3 结论
1) 通过以原位生成的邻亚甲基醌中间体与3-氯吲哚啉酮为底物,在碱性条件下发生Michael加成/环化串联反应,高收率、高非对映选择性地构建了双杂环螺环化合物。
2) 该体系条件温和,操作简便,经济性好,底物范围较广。对于其不对称合成有待进一步研究。
[1] TAN Bin, CANDEIAS N R, BARBAS III C F. Construction of bispirooxindoles containing three quaternary stereocentres in a cascade using a single multifunctional organocatalyst[J]. Nature Chemistry, 2011, 3(3): 473−477.
[2] MUGISHIMA T, TSUDA M, WATANABE M, et al. Absolute stereochemistry of citrinadins A and B from marine-derived fungus[J]. The Journal of Organic Chemistry, 2005, 70(23): 9430−9435.
[3] GALLIFORD C V, SCHEIDT K A. Pyrrolidinyl -spirooxindole natural products as inspirations for the development of potential therapeutic agents[J]. Angewandte Chemie Intemational Edtion, 2007, 46(46): 8748−8758.
[4] JANIN Y L. Antituberculosis drugs:ten years of research[J]. Bioorganic & Medicinal Chemistry, 2007,15(7): 2479−2513.
[5] KUHEN K L, ELLIS Y H, BURSULAYA B, et al. Oxindoles with anti-HIV activity: US 2004/037247A1[P]. 2004−02−26.
[6] PAULS H W, SAMPSON P B, FORREST B T, et al. Plk-4 inhibitors and methods of treating cancer with same: US 2012/048411A1[P]. 2012−03−01.
[7] DOU Xiaowei, YAO Weijun, ZHOU Bo, et al. Asymmetric synthesis of 3-spirocyclopropyl-2-oxindoles via intramolecular trapping of chiralaza-ortho-xylylene[J]. Chemical Communications, 2013, 49(80): 9224−9226.
[8] DOU Xiaowei, ZHOU Bo, YAO Weijun, et al. A facile approach for the asymmetric synthesis of oxindoles with a 3‑sulfenyl-substituted quaternary stereocenter[J]. Organic Letters, 2013, 15(19): 4920−4923.
[9] NOOLE A, JARVING I, WERNER F, et al. Organocatalytic asymmetric synthesis of 3-chlorooxindoles bearing adjacent quaternary-tertiary centers[J]. Organic Letters, 2012, 14(18): 4922−4925.
[10] LI Junhua, FENG Tingfan, DU Daming. Construction of spirocyclopropane-linked heterocycles containing both pyrazolones and oxindoles through Michael/alkylation cascade reactions[J]. The Journal of Organic Chemistry, 2015, 80(22): 11369−11377.
[11] HSIAO C C, RAJA S, LIAO H H, et al. Ortho-quinone methides as reactive intermediates in asymmetric brønsted acid catalyzed cycloadditions with unactivated alkenes by exclusive activation of the electrophile[J]. Angewandte Chemie Intemational Edtion, 2015, 54(19): 5762−5765.
[12] ALAMSETTI S K, SPANKA M, SCHNEIDER C. Synergistic Rhodium/phosphoric acid catalysis for the enantioselective addition of oxonium ylides to-quinone methides[J]. Angewandte Chemie Intemational Edtion, 2016, 55(7): 2392−2396.
[13] CHEN Pin, WANG Kai, GUO Wengang, et al. Enantioselective reactions of 2-sulfonylalkyl phenols with allenic esters: dynamic kinetic resolution and [4+2] cycloaddition involving- quinone methide intermediates[J]. Angewandte Chemie Intemational Edtion, 2017, 56(13): 3689−3693.
[14] WU Bo, YU Zhaoyuan, GAO Xiang, et al. Regioselective-addition of deconjugated butenolides: enantioselective synthesis of dihydrocoumarins[J]. Angewandte Chemie Intemational Edtion, 2017, 56(14): 4006−4010.
[15] ZHANG Jianlin, LIN Lili, HE Changqiang, et al. A chiral scandium-complex-catalyzed asymmetric inverse-electron- demand oxa-Diels–Alder reaction of o-quinone methides with fulvenes[J]. Chem Commun, 2017, 54(1): 74−77.
[16] ZHOU Ding, YU Xueting, ZHANG Jian, et al. Organocatalytic asymmetric formal [4+2] cycloaddition of in situ oxidation- generated-quinone methides and aldehydes[J]. Organic Letters, 2018, 20(1): 174−177.
[17] MAJUMDAR N, KORTHALS K A, WULFF W D. Simultaneous synthesis of both rings of chromenes via a benzannulation/o-quinone methide formation/electrocycli zation cascade[J]. Journal of The American Chemical Society, 2012, 134(2): 1357−1362.
[18] LIAO Daohong, LI Houhua, LEI Xiaoguang. Efficient generation of ortho-quinone methide: application to the biomimetic syntheses of (±)-schefflone and tocopherol trimers[J]. Organic Letters, 2012, 14(1): 18−21.
[19] TAKAO K, NOGUCHI S, SAKAMOTO S, et al. Total synthesis of (+)-cytosporolide A via a biomimetic hetero-Diels-Alder reaction[J]. Journal of The American Chemical Society, 2015, 137(50): 15971−15977
[20] CAO Liangliang, WANG Duosheng, JIANG Guofang, et al. An efficient route to 2, 3-disubstituted indoles via reductive alkylation using H2as reductant[J]. Tetrahedron Letters, 2011, 52(22): 2837−2839.
[21] CAO Liangliang, YE Zhishi, JIANG Guofang, et al. Rhodium-catalyzed addition of boronic acids to vinylogous imines generated in situ from sulfonylindoles[J]. Advanced Synthesis & Catalysis, 2011, 353(18): 3352−3356.
[22] LUO Jing, WU Bo, CHEN Muwang, et al. The concise synthesis of spiro-cyclopropane compounds via the dearomatization of indole derivatives[J]. Org Lett, 2014, 16(10): 2578−2581.
[23] GU Zheng, TANG Yao, JIANG Guofang. Combined di-tert-butyl peroxide and inorganic base promoted α-alkylation of ethers with arenesulfonylindoles[J]. The Journal of Organic Chemistry, 2017, 82(10): 5441−5448.
[24] WANG Ciping, JIANG Guofang. An efficient method based on indoles for the synthesis of isatins by taking advantage of I2O5as oxidant[J]. Tetrahedron Letters, 2017, 58(18): 1747−1750.
[25] ZHOU Ji, WANG Maolin, GAO Xiang, et al. Bifunctional squaramide-catalyzed synthesis of chiral dihydrocoumarins via ortho-quinone methides generated from 2-(1-tosylalkyl)phenols[J]. Chemical Communications, 2017, 53(25): 3531−3534.
[26] ZHOU Ji, HUANG Wenjun, JIANG Guofang. Synthesis of chiral pyrazolone and spiropyrazolone derivatives through squaramide-catalyzed reaction of pyrazolin-5-ones with o-quinone methides[J]. Organic Letters, 2018, 20(4): 1158−1161.
(编辑 陈灿华)
Synthesis of spirooxindoles through [4+1] cycloaddition of-quinone methides with 3-chlorooxindoles
DU Shenghua1, 2, ZHOU Ji2, JIANG Guofang2
(1. Hunan Research Institute of Chemical Industry Co. Ltd., Changsha 410014, China; 2. College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China)
The spirooxindoles were synthesized in high yield with excellent diastereoslectivities using 3-chlorooxindoles and-quinone methides (o-QMs) as materials through the Michael addition/cyclization cascade process under basic condition. To get the two spirocyclic compound involving benzofuran and oxindole skeletons under mild and easy-handle conditions,3-chlorooxindoles was introduced to react with the o-QMs, which were synthesizedfrom 2-(1-tosylalkyl) phenols. And the optimized target products were obtained through condition selection. The gram-scale experiments were conducted to verify the practicality of this method. The results show that under optimized condition, the yield of target product can achieve 92% and the diastereoselectivityris more than 20:1. And high yield and diastereoselectivity can be maintained when the substrate amount increases. The method can be applied widely and multiple compounds such as 2-(1-tosylalkyl) phenols and 3-chlorooxindoles can be used as materials in this method.
3-chlorooxindole;-quinone methides; Michael addition/cyclizaiton; spirooxindoles
10.11817/j.issn.1672-7207.2018.05.006
0626.2
A
1672−7207(2018)05−1062−10
2018−01−10;
2018−03−02
国家自然科学基金资助项目(51578224) ( Project(51578224) supported by the National Natural Science Foundation of China)
江国防,博士,教授,从事有机合成研究;E-mail: gfjiang@hnu.edu.cn
