敲除LSD1基因对人慢性髓系白血病K562细胞周期的影响
2018-05-24任思蕊王冰蕊冯甜甜石莉红
任思蕊, 王冰蕊, 郭 青, 冯甜甜, 王 鼎, 高 洁, 石莉红
中国医学科学院, 北京协和医学院血液病医院(血液学研究所), 实验血液学国家重点实验室, 天津 300020
表观遗传是在DNA序列信息不变的情况下,细胞内部发生的可遗传改变,方式主要有DNA和组蛋白的修饰、染色质的重塑等[1]。表观遗传学参与了肿瘤的发生发展,在已有的白血病研究中,发现许多原癌基因和抑癌基因的启动子区域CpG岛发生异常的甲基化修饰,例如ABLl(P1)基因[2]、ECAD基因[3]、EphA3基因[4]等。但是关于组蛋白修饰参与白血病发生的研究较少。
在构成染色质的组蛋白上,可以发生甲基化、乙酰化、泛素化等多种修饰,进而调控基因表达[5]。LSD1是组蛋白去甲基酶,它催化组蛋白赖氨酸特异的去甲基反应从而完成对组蛋白的修饰,抑制靶基因的转录[6]。LSD1在哺乳动物造血系统中发挥着重要的生理作用。LSD1敲除的小鼠胚胎发育7.5d死亡,通过对LSD1条件敲除小鼠模型的研究显示,LSD1在造血发生过程中高表达,并且对红系、巨核系、粒系细胞的终末分化必不可少[7]。在AML细胞中,通过抑制LSD1的活性,降低了AML细胞的集落形成能力,并促进细胞凋亡[8],这暗示LSD1可能通过组蛋白修饰参与白血病的发展。
在我们的前期实验中,已经利用CRISPR/Cas9技术构建了人慢性髓系白血病K562的LSD1基因敲除株,发现LSD1的敲除显著抑制了CD235a的表达,并且显著减慢了K562细胞的增殖速度[9]。为了探究LSD1敲除后K562细胞增殖变慢的原因,本研究利用流式细胞仪检测细胞凋亡的改变以及细胞周期的变化,发现敲除LSD1后,不影响K562细胞凋亡水平;但是细胞周期发生明显改变,阻滞在G0/G1期的细胞变多,S期和G2/M期的细胞变少,进而导致细胞的增殖变慢。
1 材料方法
1.1 仪器与试剂
流式细胞仪LSR II(美国BD公司);K562细胞系原购自美国ATCC公司,后经由本实验室保存; K562细胞LSD1纯合敲除株(LSD1-/-)、杂合敲除株(LSD1+/-)由本实验室构建,传代并冻存;细胞培养所用的胎牛血清、L-谷氨酰胺、RPMI 1640基础培养液、青霉素-链霉素抗生素、丙酮酸钠等细胞培养相关试剂购自美国Gibco公司; 细胞凋亡检测试剂盒购于美国BD公司。
1.2 细胞培养
K562细胞使用的基础培养液为RPMI 1640,添加1%的青霉素-链霉素、1%的L-glutamine以及10%的胎牛血清,K562细胞培养条件为37℃恒温、5% CO2孵箱培养,培养密度为1×105个/mL,细胞密度达到1×106个/mL时进行细胞传代。
1.3 K562细胞凋亡检测
试剂稀释:细胞凋亡检测试剂盒中提供的5×Binding Buffer,使用无菌PBS稀释至1×Binding Buffer,4℃预冷。
K562细胞的准备:K562细胞计数取1×106个细胞,300 g离心5 min,弃上清,用PBS缓冲液清洗1遍,吸弃上清后使用100 μL 1×Binding Buffer将细胞重悬。
细胞染色:加入5 μL的Annexin V-FITC混匀后,避光,室温孵育15 min;上机检测前5 min再加入5 μL的PI染料并补加200 μL的1×Binding Buffer。
制备好的细胞悬液使用流式细胞仪LSRⅡ检测细胞凋亡比例,使用FITC和PI两个通道,检测结束后用软件FlowJo7.6.1进行分析,流式细胞仪检测显示的细胞分布在四象限内,Annexin V-/PI-为活细胞,Annexin V+/PI-和Annexin V+/PI+为早期凋亡以及晚期凋亡的细胞。
1.4 细胞周期检测
细胞固定:收集细胞并用PBS洗涤,300 g离心5 min后弃掉上清;每组细胞中加入1 mL 70%乙醇,混匀后4℃固定24 h以上。
细胞染色与检测:取固定完毕的细胞,使用PBS清洗一遍,300 g离心5 min后弃掉上清。重新加入50 μL PBS及3 μL核糖核酸酶,室温孵育10 min后加入150 μL PI的PBS溶液,室温避光染色10 min;染色后使用LSR II流式细胞仪检测,并使用ModFit程序分析G0/G1,S和G2/M期细胞比率。
1.5 统计学分析方法

2 结果与分析
2.1 LSD1基因敲除对K562细胞增殖周期的影响
前期研究中发现,敲除LSD1基因后K562细胞增殖受到显著抑制,LSD1+/-细胞株增殖速度显著慢于WT组,LSD1-/-细胞株的增殖速度与WT组相比,差异极显著[9]。为了进一步探究敲除LSD1基因后K562细胞增殖受到显著抑制的原因,本实验检测了K562细胞的增殖周期,细胞周期中的G0和G1期是DNA合成前期,进入S期后DNA开始合成,G2期DNA合成结束。将培养中的K562细胞野生型、LSD1+/-、LSD1-/-细胞株同时进行PI染色,PI可以与DNA结合,其荧光强度直接反映了细胞内DNA含量(图1A)。分析3株细胞所处细胞周期内各阶段细胞比例,结果显示敲除LSD1后,被阻滞在G1期的细胞比例升高,然而在S期的细胞比例明显降低,进入G2/M期的细胞比例也降低(图1B)。以上结果表明,敲除LSD1基因后K562细胞的增殖周期被阻滞在G0/G1期,进入DNA复制期的细胞减少,从而使进入分裂状态细胞比例减少,细胞增殖减慢。

图1 LSD1敲除对K562细胞增殖周期的影响Fig.1 Effects of LSD1 knockout on the cell cycle of K562 cells.A.PI染色以及流式细胞仪分析各组细胞的细胞周期分布比例;B.统计学分析各组细胞的细胞周期分布比例。*表示P<0.1水平差异显著,**表示P<0.01水平差异显著,***表示P<0.001水平差异显著。
2.2 LSD1基因敲除对K562细胞凋亡水平的影响
为了探究LSD1+/-和LSD1-/-细胞株增殖减慢的原因,除细胞周期改变外,是否由于敲除基因LSD1影响细胞凋亡水平所引起,本实验将培养中的K562细胞野生型、LSD1+/-、LSD1-/-细胞株同时进行细胞凋亡检测,并使用软件FlowJo7.6.1分析,Annexin V-/PI-为活细胞,Annexin V+/PI-和Annexin V+/PI+为早期凋亡以及晚期凋亡的细胞,统计分析凋亡细胞的总比率,结果显示: 敲除LSD1后,不影响K562细胞凋亡水平(图2)。
3 讨论
LSD1又名KDM1A,是胺氧化酶家族中的一员,其C端具有AOL结构域,主要包括FAD(黄素腺嘌吟二核苷酸)结合结构域和催化活性中心两个部分,在FDA的参与帮助下,LSD1可以与组蛋白底物结合,特异催化组蛋白H3K9me1/2和H3K4me1/2去甲基化[10~12]。
研究表明,组蛋白修饰在生理以及病理状态下的造血过程中均发挥核心作用。在急性白血病患者的基因组中,由于染色质修饰基因的各种后天病变,或其表达水平的变化,表观遗传学的整体状态发生明显改变[13]。在多种血液系统淋系和髓系恶性肿瘤中,组蛋白-赖氨酸-N-甲基转移酶EZH2被证明发生了突变[14];组蛋白甲基转移酶DOT1L的小分子抑制剂显示出对MLL(混合谱系白血病)的临床治愈性[15,16];另外,组蛋白去甲基转移酶KDM5A(JARID1A)、KDM5C(JARID1C)和KDM6A(UTX)在实体以及血液系统肿瘤中均出现了大规模重复性突变,因此LSD1/KDM1A也一度成为临床治疗AML的研究热点[17]。LSD1在AML(急性髓系白血病)中高表达,多种LSD1抑制剂在临床应用中展现出对AML一定的治疗作用[18],在MLL-AF9+AML细胞中,敲低LSD1的表达水平,H3K4me2/H3K4me3比例升高从而降低了白血病发展风险[19~21]。但是关于LSD1在白血病细胞分化、增殖以及凋亡的研究比较少。
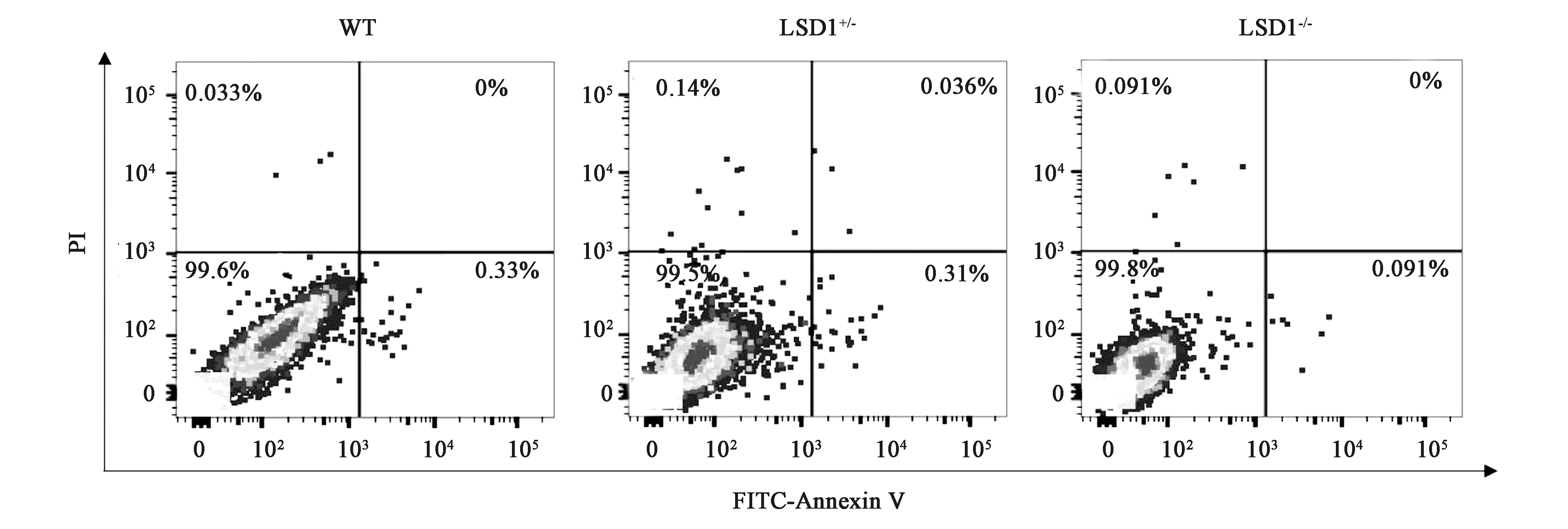
图2 LSD1基因敲除对K562细胞凋亡水平的影响Fig.2 The effect of LSD1 knockout on apoptosis level of K562 cells.
K562细胞是研究白血病发病机理的重要模型,我们在前期的实验中,已经使用人慢性髓系白血病K562细胞,利用 CRISPR/Cas9技术对K562细胞的LSD1 Exon1进行定点切割,并获得了LSD1+/-、LSD1-/-K562细胞株,通过对敲除株的研究发现,LSD1+/-、LSD1-/-K562细胞株的红系特异性表面标记CD235a的表达被显著抑制,并且与LSD1的表达呈剂量依赖关系。因此我们推测,LSD1可能参与调控了K562细胞红系分化过程中的红细胞成熟进程。同时我们发现LSD1+/-、LSD1-/-K562细胞株的增殖能力明显下降,这暗示了LSD1基因的抑制能够有助于白血病的治疗,因此本实验利用细胞凋亡检测,验证了敲除LSD1基因后,K562细胞凋亡水平并未受到影响。同时,数据显示野生型K562细胞凋亡很不明显,因此有必要进一步研究在饥饿或诱导剂处理下,LSD1对K562细胞凋亡是否有影响。另外,敲除LSD1基因后,野生型和敲除株细胞的K562细胞被阻滞在细胞周期的G0/G1期,进入DNA复制期的细胞变少,细胞增殖明显减慢;结果中细胞周期改变与LSD1敲除比例未出现明显的计量依赖性,因此认为,除了影响细胞周期外,可能还存在其他的信号通路,受到LSD1敲除的影响从而减慢了细胞的增殖。
综上所述,LSD1基因K562细胞在红系分化进程中至关重要,并且起到维持细胞凋亡平衡状态以及细胞顺利增殖的作用。因此,抑制LSD1的活性,理论上可以成为治疗白血病的策略手段。同时,本研究也为临床上LSD1抑制剂治疗白血病提供了有效的理论依据。
参 考 文 献
[1] Huang B, Jiang C, Zhang R. Epigenetics: The language of the cell? [J]. Epigenomics, 2014, 6(1): 73-88.
[2] Issa J P, Kantarjian H, Mohan A,etal.. Methylation of the ABL1 promoter in chronic myelogenous leukemia: Lack of prognostic significance[J]. Blood, 1999, 93(6): 2075-2080.
[3] Seeliger B, Wilop S, Osieka R,etal.. CpG island methylation patterns in chronic lymphocytic leukemia[J]. Leuk Lymph., 2009, 50(3): 419-426.
[4] Dottori M, Down M, Huttmann A,etal.. Cloning and characterization of EphA3(Hek) gene promoter: DNA methylation regulates expression in hematopoietic tumor cells[J]. Blood, 1999, 94(7): 2477-2486.
[5] Deans C, Maggert K A. What do you mean, Epigenetic? [J]. Genetics, 2015, 199(4):887-896.
[6] Shi Y, Lan F, Matson C,etal.. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1[J]. Cell, 2004, 119(7): 941-953.
[7] Wang J, Scully K, Zhu X,etal.. Opposing LSD1 complexes function in developmental gene activation and repression programmes[J]. Nature, 2007, 446(7138): 882-887.
[8] Daniel P M, Alison E M, Daniel H W,etal.. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date[J]. Med. Res. Rev., 2015, 35(3):586-618.
[9] 高 洁, 任思蕊, 王冰蕊,等. 利用CRISPR/CAS9技术敲除LSD1基因显著抑制人慢性髓系白血病K562细胞的增殖与CD235a的表达[J]. 中国实验血液学杂志, 2017, 25(5): 1327-1333.
[10] Rotili D, Mai A. Targetinghistone demethylases: A new avenue for the fight against cancer[J]. Genes Cancer, 2011, 2(6): 663-679.
[11] Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes[J]. J. Biol. Chem., 2007, 282(49): 35425-35429.
[12] Chen Y, Yang Y, Wang F,etal.. Ciystal structure of human his-tonelysine-specific demethylase 1 ( LSD1) [J]. Proc. Natl. Acad. Sci. USA, 2006, 103(38): 13956-13961.
[13] Ly P V, Luisa L, Stephen D N. Histone-modifying enzymes: Their role in the pathogenesis of acute leukemia and their therapeutic potential[J]. Int. J. Hematol., 2013, 97(2):198-209.
[14] Dawson M A, Kouzarides T. Cancer epigenetics: from mechanism to therapy[J]. Cell, 2012, 150(1): 12-27.
[15] Zhang K, Dent S Y. Histone modifying enzymes and cancer: going beyond histones[J]. J. Cell Biochem., 2005, 96(6): 1137-1148.
[16] Krivtsov A V,Armstrong S A. MLL translocations, histone modifications and leukaemia stem-cell development[J]. Nat. Rev. Cancer, 2007,7(11):823-833.
[17] Barretina J, Caponigro G,Stransky N,etal.. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity[J]. Nature, 2012, 483(7391): 603-607.
[18] Berglund L, Björling E, Oksvold P,etal.. A genecentric human protein atlas for expression profiles based on antibodies[J]. Mol. Cell Proteom., 2008,7(10): 2019-2027.
[19] Harris W J, Huang X, Lynch J T,etal.. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells[J]. Cancer Cell, 2012, 21(4): 473-487.
[20] Lokken A A, Zeleznik-Le N J. Breaking the LSD1/KDM1A addiction: Therapeutic targeting of the epigenetic modifier in AML[J]. Cancer Cell, 2012,21(4): 451-413.
[21] Schenk T, Chen W C, Göllner S,etal.. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia[J]. Nat. Med., 2012, 18(4): 605-611.
