hTNFR1在大肠杆菌中可溶性表达及纯化
2018-05-24盖园明朱蓓薇张大伟
韩 平, 盖园明, 朱蓓薇*, 张大伟*
1.大连工业大学食品学院, 辽宁 大连 116034;2.中国科学院天津工业生物技术研究所, 天津 300308
肿瘤坏死因子(TNF-α)是一种由巨噬细胞[1]产生的具有“双面作用”的细胞因子。与其他细胞因子一样,TNF-α主要通过与细胞膜上的受体特异性结合从而发挥它的生物活性。一方面,TNF-α具有很强的抗肿瘤活性,可以特异性的识别和致死肿瘤细胞。因此,它有望成为治疗癌症和感染性免疫休克的潜在药物[2,3]。另一方面,如果TNF-α在机体内过度表达[4,5],会导致多种炎症疾病的发生,如类风湿关节炎等。为了进一步促使TNF-α被广泛应用于临床医学研究和癌症治疗研究,常常需要人为的干扰或者阻断TNF-α在细胞内的部分信号传导或者特异性的进行体外注射来达到研究和治疗的目的。
在人体内,TNF-α受体分为Ⅰ型(TNFR1)和Ⅱ型(TNFR2)两种,与TNFR2相比,TNFR1胞浆区含有转导细胞死亡信号所必需的死亡结构域[6,7],目前有多篇研究报道认为,肿瘤细胞的凋亡路径也主要由TNFR1介导完成。TNFR1的胞外区在TNF转换酶的作用下从细胞膜上[8]解离下来,它能与TNF-α结合,但不介导TNF-α的信号传导,为此可溶性TNFR1常常被选作TNF-α的抑制剂,用于减少TNF-α对细胞的毒性,而同时和TNF-α联合应用于疾病的治疗。
可溶性hTNFR1蛋白富含半胱氨酸[9],易形成二硫键结构,但大肠杆菌合成二硫键的能力有限,易形成大量包涵体。本实验旨在通过融合标签技术[10]和共表达分子伴侣的策略来辅助hTNFR1蛋白正确折叠,从而提高hTNFR1蛋白在大肠杆菌的可溶性表达。同时,通过对实验结果的分析,为后续完善BL21(DE3)pET系统提供新思路。
1 材料与方法
1.1 材料
1.1.1质粒与菌株 本文所用菌株和质粒均列于表1,构建质粒所用引物均列于表2,引物用Oligo软件进行设计,送金唯智生物科技有限公司合成。
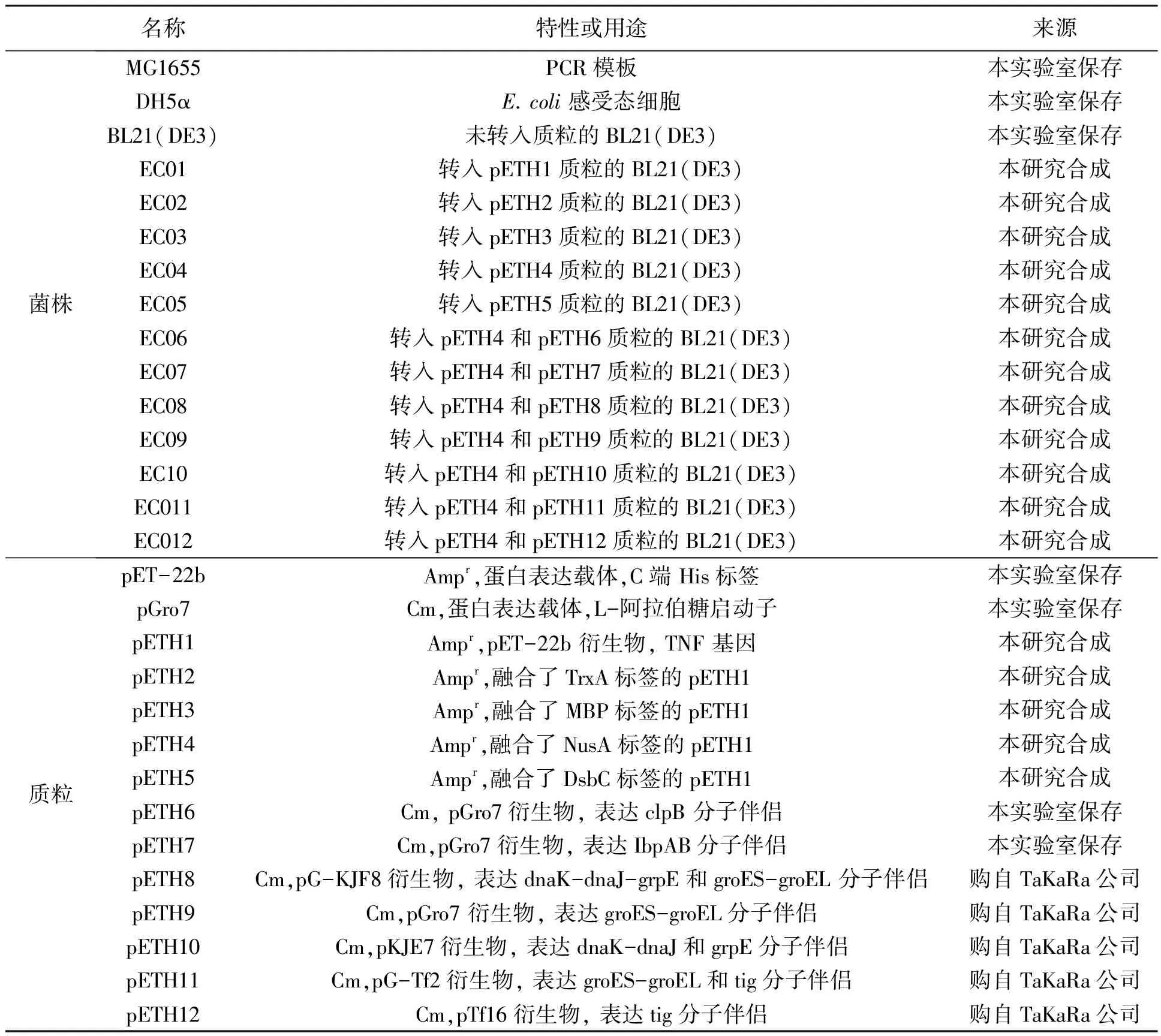
表1 本研究所用菌株与质粒Table 1 Strains and plasmids used in this study.

表2 本研究所用引物Table 2 Primers used in this study.
1.1.2主要试剂 彩虹广谱蛋白Maker、异丙基-β-d-硫代半乳糖苷(isopropyl-β-dthiogalactoside,IPTG)、TEV蛋白酶、蛋白酶抑制剂Cocktail和考马斯亮蓝R250购于北京索莱宝生物科技有限公司;PrimerSTAR Mix 购自TaKaRa公司;2×TaqPCR MasterMix、DNA Marker(DP302-02) 购于北京天根生化科技有限公司;细菌基因组DNA提取试剂盒、质粒小提试剂盒(D6943-02)和柱式PCR产物纯化试剂盒(D6492-02)购自Omega公司;Gibson Assembly 1.2 Master Mix为纽英伦生物技术有限公司产品;鼠抗His单克隆抗体(一抗)和辣根过氧化物酶标记的羊抗鼠IgG抗体(二抗)购于北京中杉金桥生物技术有限公司;其余试剂均为进口或国产分析纯。
1.2 方法
1.2.1重组质粒的构建及验证 根据NCBI中hTNFR1的基因序列为模板(登录号:1NCF_A),将其提供给金唯智生物科技有限公司进行全基因合成和密码子优化。以合成的hTNFR1基因为模板,TNFR1-F和TNFR1-R为引物,进行PCR扩增。PCR反应体系为:20 μL Prime STAR Max Premix(2×),1 μL DNA模板,2 μL 10 μmol/L正向引物和2 μL 10 μmol/L反向引物,加16 μL ddH2O至总体积为40 μL。PCR反应程序:98℃ 10 min;98℃ 30 s,55℃ 15 s,72℃ 1 min/2 kb,30个循环;72℃ 3 min,4℃保存。扩增出的片段用1%琼脂糖凝胶电泳鉴定,验证后的PCR产物进行柱回收。将回收后的hTNFR1片段和pET-22b载体进行Gibson Assembly[11]。连接后的产物转化到E.coli感受态细胞DH5α中,用含有Amp(100 μg/mL)抗生素的LB平板进行阳性克隆筛选,从而获得重组质粒pETH1。带有融合标签TrxA、MBP、NusA和DsbC的质粒pETH2、pETH3、pETH4和pETH5也用此方法进行构建,流程参考图1。
挑取转化子进行菌落PCR验证,单菌落溶于10 μL无菌水中,取2 μL菌液为PCR模板,TNFR1-F和TNFR1-R为验证引物,用2×Taq酶进行PCR扩增,PCR产物用1%琼脂糖凝胶电泳鉴定。
1.2.2重组蛋白的诱导表达和SDS-PAGE分析 将pETH1电转化到感受态细胞BL21(DE3)中,转化产物涂布到含有Amp(100 μg/mL,下同)抗生素的LB平板,放置于37℃恒温培养箱过夜培养。挑取单菌落接种于含Amp抗生素的LB试管中,37℃过夜培养,菌液按1∶100转接到装有30 mL含Amp抗生素的TB培养基的250 mL摇瓶,37℃ 220 r/min震荡培养至OD600达到2.0时,加入IPTG(终浓度为0.5 mmol/L)进行诱导表达,25℃ 200 r/min低温诱导表达20 h。摇瓶发酵结束后,制备胞内全菌、上清和沉淀三组分的蛋白样品,具体操作步骤为:先测菌生长OD600,取1 mL菌液,4℃ 4 000 r/min离心10 min,收集菌体。用Tris-HCl缓冲液(pH 8.0)将菌体OD600统一稀释到4.0, 取990 μL的菌液加入10 μL蛋白酶抑制剂充分混匀后,进行超声破碎后,沸水煮样15 min,13 000 r/min离心4 min,12% SDS-PAGE分析胞内蛋白表达。
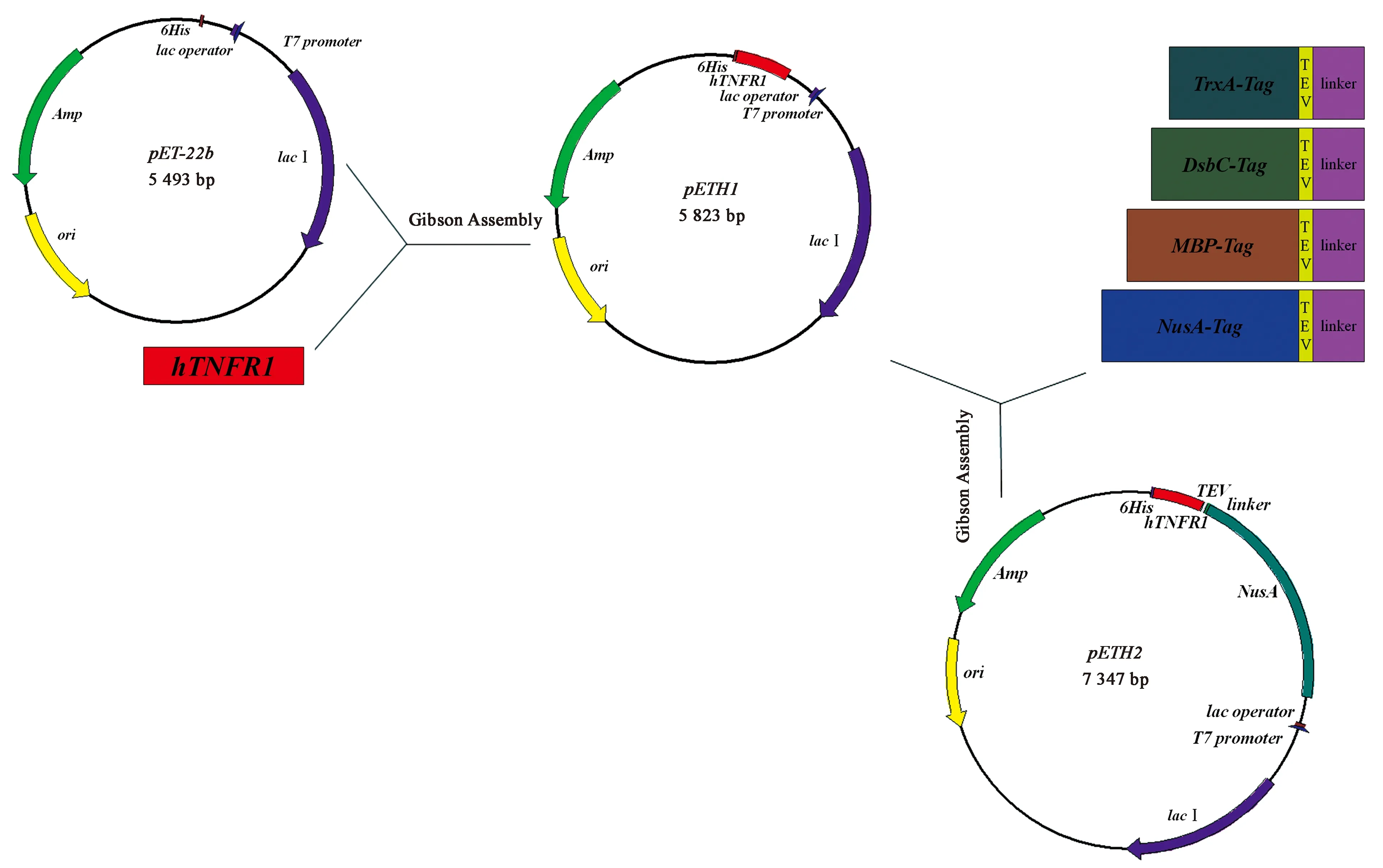
图1 重组质粒的构建Fig.1 Construction of recombinant plasmids.
1.2.3分子伴侣共表达体系的构建和诱导表达 将pETH2重组质粒分别和7种分子伴侣质粒(pETH6、pETH7、pETH8、pETH9、pETH10、pETH11、pETH12)共转化到感受态细胞BL21(DE3)中,将转化产物涂布到含有Amp和Cm(34 μg/mL,下同)抗性的LB平板上,从而筛选出阳性转化子,获得同时含有pETH2和分子伴侣质粒的工程菌株。
挑取单菌落接种于含有Amp和Cm抗生素的LB试管中,37℃过夜培养后,菌液按1∶100转接到摇瓶中,并且加入四环素(终浓度为5 ng/mL和20 ng/mL)或L-阿拉伯糖(终浓度为0.05%和0.2%)以诱导分子伴侣表达(pETH11 用四环素诱导,pETH6、pETH7、pETH9、pETH10和pETH12 用L-阿拉伯糖诱导,pETH8用四环素和L-阿拉伯糖诱导),37℃ 220 r/min震荡培养至OD600达到2时,加入IPTG(终浓度0.5 mmol/L),25℃ 200 r/min低温诱导表达20 h,制备蛋白样品。以转入重组质粒pETH2的EC04菌株的发酵结果为阴性对照,12%的SDS-PAGE分析胞内蛋白表达。
1.2.4重组蛋白的纯化 取10 mL培养菌液,4℃ 5 000 r/min离心10 min收集菌体,加入菌体裂解液重悬菌体进行超声破碎,4℃ 13 000 r/min离心5 min收集上清液。取1 mL Ni-NTA填料于柱子中,用10倍柱体积的蒸馏水和washing buffer(30 mmol/L咪唑,pH 8.0)先后清洗柱子,增强填料与蛋白的结合程度。将填料取出与蛋白裂解液混合,冰上结合2 h,用10~15倍柱体积的washing buffer洗去杂蛋白,最后加入1mL Elution buffer (500 mmol/L咪唑,pH 8.0)进行一步洗脱,纯化蛋白用12%SDS-PAGE鉴定。
1.2.5TEV蛋白酶酶切重组蛋白 对纯化蛋白进行酶切实验,去除NusA标签。反应体系为:酶切蛋白量为100 μL,酶用量依次为0.5 μL、1 μL、2 μL、5 μL,10℃在1×TEV Buffer中反应6 h。另一组4℃酶切过夜,反应体系如上。待反应结束后,制备蛋白样品,沸水煮样15 min,13 000 r/min离心5 min,用12%SDS-PAGE鉴定。
1.2.6重组蛋白Western blot鉴定 将蛋白样品先进行12%SDS-PAGE电泳,再经湿法转膜2 h,将蛋白转到PVDF膜上,用5%的脱脂奶粉封闭1 h,放置于含鼠抗His单克隆抗体(一抗)的TBS/T(1∶2 000稀释)中孵育45 min,用TBS/T洗膜3次,每次5 min,再放置于含有辣根过氧化物酶标记的羊抗鼠IgG抗体(二抗)的TBS/T(1∶2 000稀释)中反应1 h,先用TBS/T洗膜3次,再用TBS洗膜1次,最后用DAB显色试剂盒显色。
2 结果与分析
2.1 重组质粒pETH1的鉴定
重组质粒pETH1的验证结果见图2,约在330 bp处有明亮单一的条带,与目的条带大小一致。将剩余菌液接种于试管中进行过夜培养,提质粒,将质粒送予金唯智生物科技有限公司进行测序,测序结果用DNA Man软件进行比对。从而获得带有hTNFR1基因片段的重组质粒pETH1。

图2 重组质粒pETH1的菌落验证Fig.2 Colony PCR verification of recombinant plasmid pETH1.M:DNA Maker Ⅲ;1~2:PCR扩增产物; CK:阴性对照.
2.2 重组蛋白的诱导表达和SDS-PAGE分析
测定工程菌EC01在37℃下的菌体生长曲线,结果见图3。表达的目的蛋白大小为12.4 kDa,结果显示目的蛋白全部以包涵体的形式存在于沉淀中(图4)。分析原因可能因为大肠杆菌胞内合成二硫键能力有限,目的蛋白中的二硫键结构没有正确形成,致使目的蛋白不能正确折叠,从而形成大量包涵体。
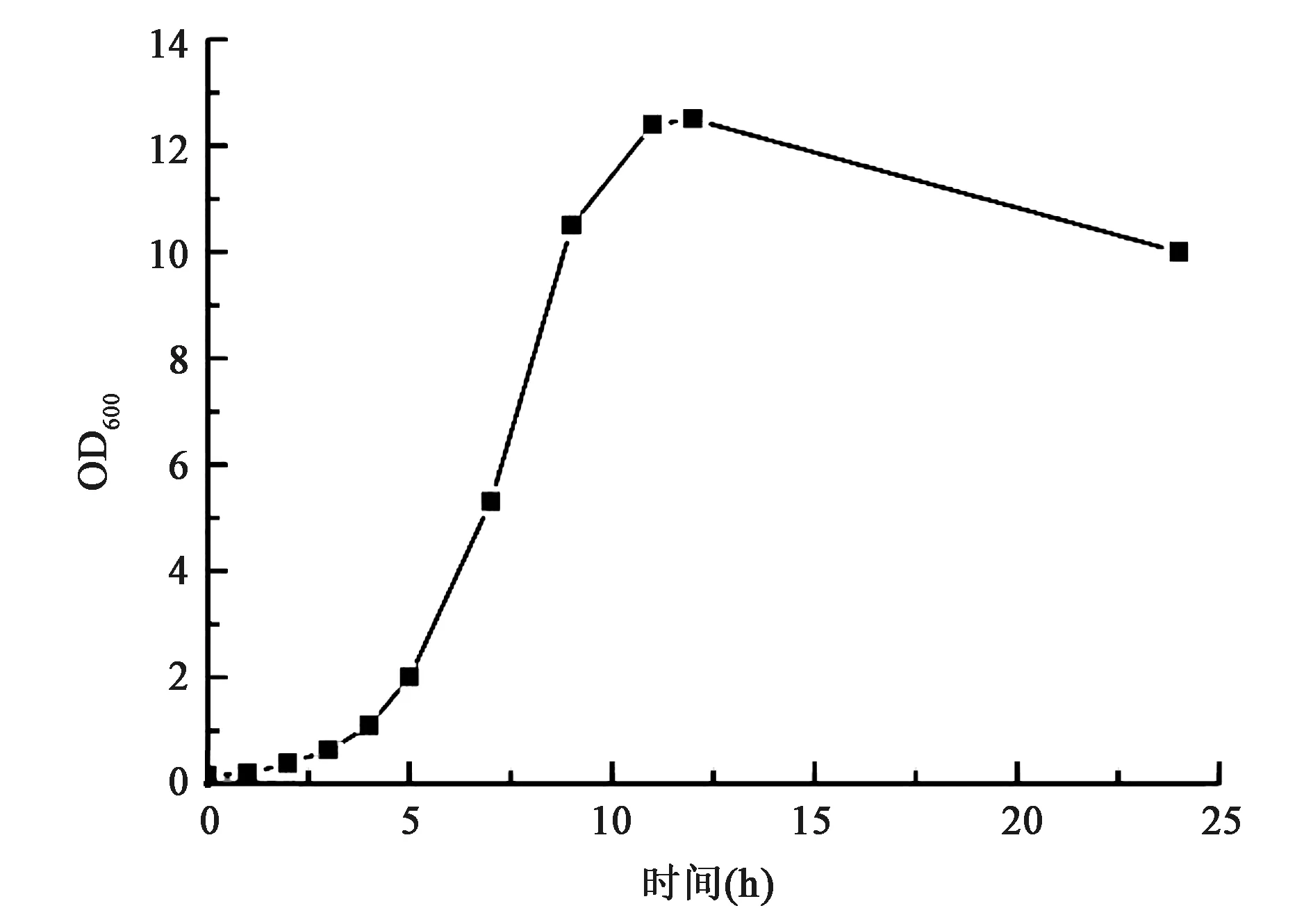
图3 EC01菌株生长曲线Fig.3 The growth curve of EC01 strain.
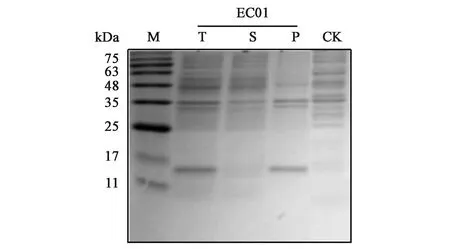
图4 SDS-PAGE 检测重组蛋白表达情况Fig.4 SDS-PAGE analysis of recombinant protein expression.M:蛋白 Maker;T:总蛋白;S:可溶性蛋白;P:沉淀;CK:转入pET-22b质粒的BL21(DE3)
2.3 融合蛋白的诱导表达和SDS-PAGE分析
为了提高目的蛋白的可溶性表达,在重组质粒pETH1基础上,在hTNFR1蛋白的N端融合TrxA、MBP、NusA和DsbC 4个在大肠杆菌表达系统中适用的蛋白标签,构建了重组质粒pETH2、pETH3、pETH4和pETH5。将其分别电转入感受态细胞BL21(DE3)中,形成了4株工程菌EC02、EC03、EC04和EC05。将4株菌株发酵并制备蛋白样品,用12%SDS-PAGE鉴定,结果见图5。EC04菌株的可溶性组分在相对分子量大小为68 kDa处有明显条带,与NusA-sTNFR1融合蛋白大小一致。因此,目的蛋白在融合NusA标签(EC04)后,hTNFR1的可溶性表达有少量提高,约占蛋白总表达量的20%。NusA标签的效果优于其他3种标签。
2.4 融合蛋白和分子伴侣共表达系统的SDS-PAGE分析
为了在融合蛋白的基础上进一步提高hTNFR1的可溶性表达量,构建分子伴侣与融合蛋白的共表达系统,本研究选用了7种分子伴侣质粒(pETH6、pETH7、pETH8、pETH9、pETH10、pETH11和pETH12)分别和pETH4质粒共转入感受态细胞BL21(DE3)中,形成工程菌EC06、EC07、EC08、EC09、EC10、EC11、EC12。将7株工程菌发酵培养并制备蛋白样品,用12%SDS-PAGE鉴定,结果见图6。以EC04菌株摇瓶发酵的可溶性表达量为对照,结果显示:当L-ara诱导浓度为0.2%,EC12菌株中目的蛋白可溶性表达量明显高于EC04菌株,几乎没有包涵体生成。分子伴侣tig(EC12)对hTNFR1可溶性表达有明显的促进作用。使可溶性蛋白约占总体表达量的90%。
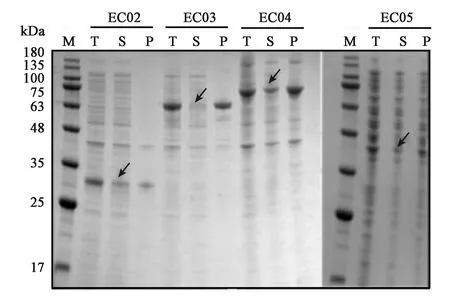
图5 SDS-PAGE 检测融合蛋白表达情况Fig.5 SDS-PAGE analysis of fusion protein expression.注:箭头指融合不同标签后的目标蛋白。M:蛋白 Maker;T:总蛋白;S:可溶性蛋白;P:沉淀
2.5 可溶性蛋白Ni-NTA纯化
将EC12菌株进行摇瓶发酵培养,可溶性表达产物经Ni-NTA纯化,将纯化过程中的各个组分用12%SDS-PAGE鉴定,结果见图7,纯化后得到了单一的目的蛋白条带。但是在流穿液中有部分目的蛋白损失,可能因为目的蛋白与Ni柱结合的不充分,后期纯化可增加Ni柱与蛋白的结合时间或适当增加Ni柱体积。
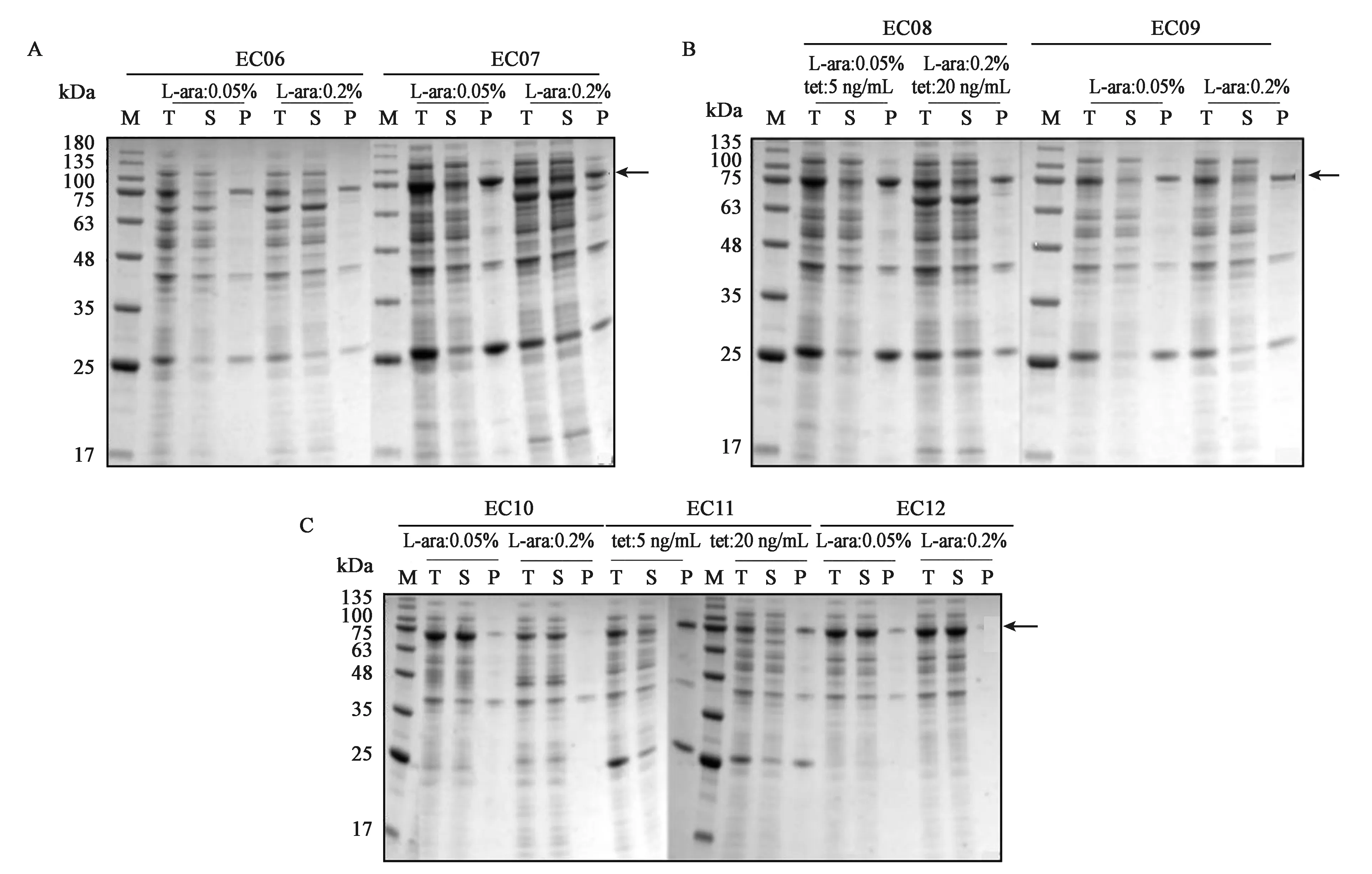
图6 SDS-PAGE 检测融合蛋白和分子伴侣共表达情况Fig.6 SDS-PAGE analysis of fusion protein and molecular chaperone co-expression system.注:箭头指融合NusA标签的目标蛋白。M:蛋白 Maker;T:总蛋白;S:可溶性蛋白;P:沉淀;L-ara:阿拉伯糖;tet:四环素

图7 SDS-PAGE 检测可溶性蛋白纯化Fig.7 SDS-PAGE analysis of soluble protein was purified.注:箭头指纯化后的目标蛋白。CK:未被IPTG诱导的蛋白;M:蛋白Maker;T:总蛋白;S:可溶性蛋白;P:沉淀
2.6 TEV蛋白酶切去除融合标签
对纯化后的蛋白进行TEV酶切,去除N端NusA标签。通过SDS-PAGE鉴定酶切效果,结果见图8。在相对分子量为55 kDa处有明显的蛋白条带,与NusA标签蛋白大小一致。在相对分子量为12.4 kDa处有蛋白条带,即为目的蛋白hTNFR1,证实大部分融合蛋白已被酶切完全。当TEV酶的用量在0.5 μL时,就可以将大部分的蛋白酶切,酶切效果较好。在4℃和10℃下进行酶切,都没有导致目的蛋白发生大量降解的情况。当TEV酶过量时,酶切过程中产生了许多杂蛋白。酶切后hTNFR1的C端有6His-tag,通过Western blot分析鉴定酶切蛋白,挑选了TEV酶为0.5 μL,10℃酶切6 h的蛋白样品进行Western blot实验,结果见图9,证实酶切后的蛋白即为hTNFR1。
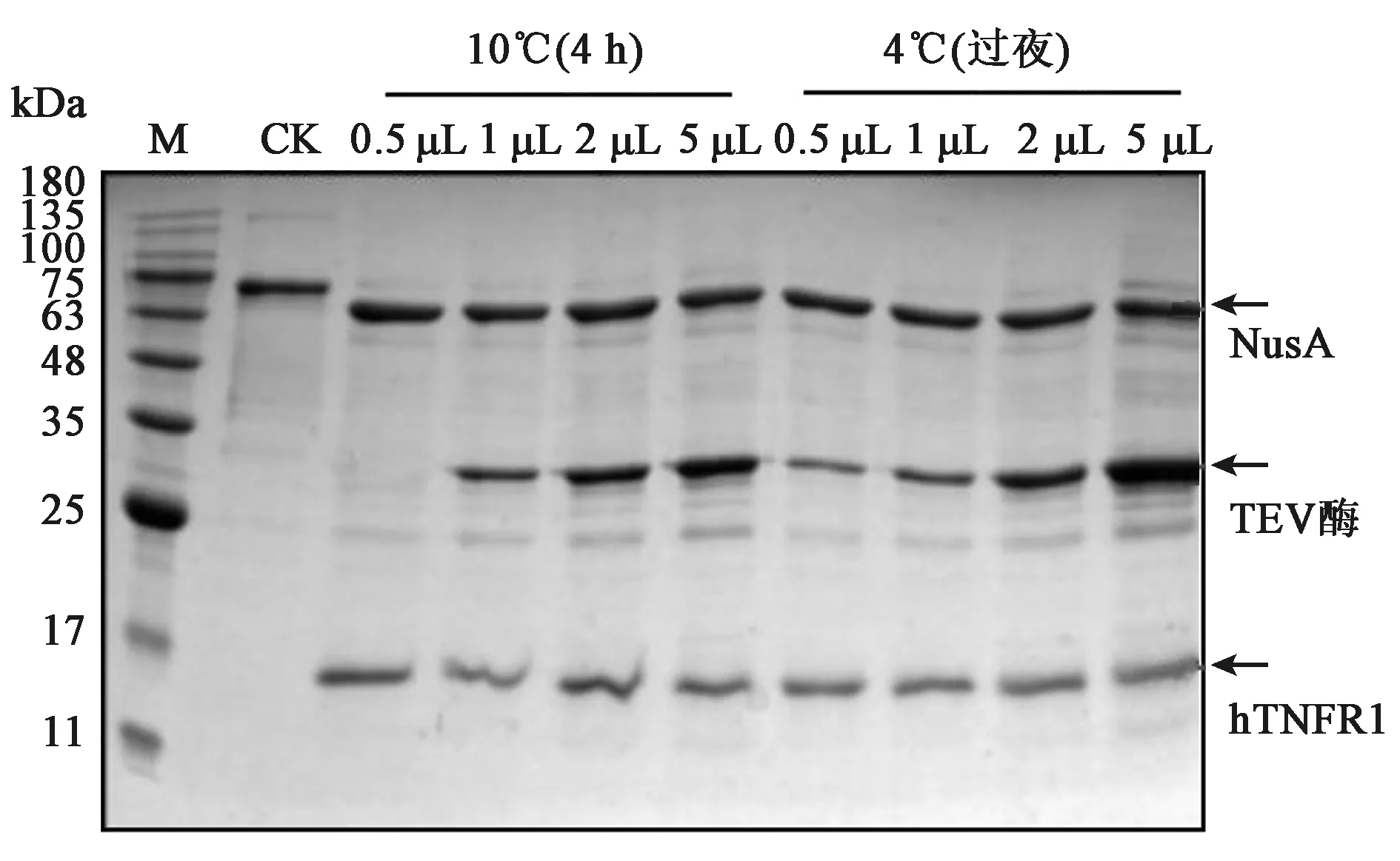
图8 SDS-PAGE检测TEV蛋白酶酶切纯化蛋白Fig.8 SDS-PAGE analysis of fusion protein that was enzymatic hydrolysis with TEV protease.注:CK:未被TEV蛋白酶酶解的融合蛋白;M:蛋白Marker
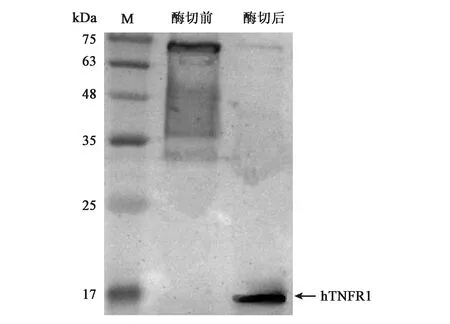
图9 Western blot鉴定酶切结果Fig.9 Western blot analysis of enzymatic hydrolysis.注:M:蛋白Marker
3 讨论
可溶性hTNFR1在临床医学应用中具有重要的意义。它可以竞争性阻断TNF与细胞膜上TNFR1的结合,从而降低TNF对细胞的毒性。因此,可溶性hTNFR1有望成为诊断[12]和治疗由TNF引起的炎症疾病的药物。要想实现hTNFR1产业化应用,获得大量高纯度hTNFR1是前提条件。单单依靠化学合成[13,14]等方法直接获取并不现实,因此可以利用基因工程大量生产hTNFR1。
安乃莉等[15]采用了在目的蛋白N端融合TrxA标签的方法,实现了可溶性hTNFR1在大肠杆菌BL21(DE3)的重组表达,总体表达量很高,但大部分目的蛋白形成了包涵体,后续需要经过复杂的变性和复性等步骤才能获得活性蛋白,大大提高了生产成本。傅蕾等[16,17]采用在目的蛋白上融合MBP标签,实现了可溶性hTNFR1在大肠杆菌JM109菌株中的重组表达并进行了蛋白纯化和活性鉴定,获得了具有活性的高纯度sTNFR1-MBP融合蛋白。本研究在以往研究基础上进一步实现hTNFR1在大肠杆菌中的高效可溶性表达。
首先,应用BL21(DE3)pET系统来重组表达可溶性hTNFR1蛋白,发酵结果显示,表达产物均以包涵体的形式存在。首先,采用融合标签技术提高hTNFR1在BL21(DE3)中的可溶性表达,其中,DsbC作为大肠杆菌周质腔中二硫键形成的异构酶,它可能具有辅助可溶性hTNFR1蛋白中二硫键形成的作用。此外,有研究报道,NusA标签可以提高重组蛋白在原核细胞中的可溶性表达,因此,选择了DsbC、TrxA、MBP和NusA标签,结果显示,融合NusA标签后,SDS-PAGE凝胶电泳分析鉴定,可溶性hTNFR1蛋白的可溶性表达量提高了约20%。
为了进一步提高hTNFR1蛋白的可溶性表达,在NusA-hTNFR1的基础上共表达大肠杆菌胞内的7种分子伴侣质粒。经SDS-PAGE凝胶电泳分析鉴定,筛选出tig分子伴侣对hTNFR1的可溶性表达有明显的促进作用。使可溶性hTNFR1蛋白的可溶性表达量提高了90%,通过Ni-NTA亲和层析纯化可溶性蛋白以及TEV蛋白酶酶切去除NusA标签后,Western blot检测酶切后的蛋白,在分子量约12.4 kDa 的位置有蛋白条带且与可溶性hTNFR1蛋白理论大小相一致,因此,结果表明本研究实现了可溶性hTNFR1在BL21(DE3)菌株中的高效可溶性表达,获得了大量高纯度的hTNFR1,为后续hTNFR1在临床医学方面的研究奠定了良好的基础。
参 考 文 献
[1] Desplatjégo S, Burkly L, Putterman C. Targeting TNF and its family members in autoimmune/inflammatory disease[J]. Mediat. Inflamm., 2015, 2014(2014):628748.
[2] Sidhu R S, Bollon A P. Tumor necrosis factor activities and cancer therapy——a perspective[J]. Pharmacol. Therapeut., 1993, 57(1):79-128.
[3] Martínez-Reza I, Díaz L, García-Becerra R. Preclinical and clinical aspects of TNF-α and its receptors TNFR1 and TNFR2 in breast cancer[J]. J. Biomed. Sci., 2017, 24(1):90.
[4] Taguchi T. Phase I study of recombinant human tumor necrosis factor (rHu-TNF:PT-050).[J]. Cancer Chem. Pharm., 1989, 29(2):144-150.
[5] Himmerich H, Fulda S, Linseisen J,etal.. TNF-alpha, soluble TNF receptor and interleukin-6 plasma levels in the general population.[J]. Eur. Cytok. Network, 2006, 17(3):196-201.
[6] 黄建生, 王昌才, 郭明秋. 肿瘤坏死因子受体的生理作用及在疾病检测中的意义[J]. 国际检验医学杂志, 1997(3):110-113.
[7] Chen C H. Structural and functional analysis of the two human tumor necrosis factor receptors[D]. USA: Thomas Jefferson University, Master Dissertation, 1997.
[8] Rothe J, Gehr G, Loetscher H,etal.. Tumor necrosis factor receptors-structure and function[J]. Immunol. Res., 1992, 11(2):81-90.
[9] He F, Dang W, Saito K,etal.. Solution structure of the cysteine-rich domain in Fn14, a member of the tumor necrosis factor receptor superfamily[J]. Protein Sci., 2009, 18(3):650-656.
[10] Zblewska K, Krajewska J, Zolkiewski M,etal.. Role of the disaggregase ClpB in processing of proteins aggregated as inclusion bodies [J]. Archiv. Biochem. Biophys., 2014, 555-556(8):23-27.
[11] Isothermal Reaction (Gibson Assembly) 11Master Mix [J]. Cold Spring Harb. Protoc., 2017, doi:10.1101/pdf.reco90019.
[12] Li M. Expression of soluble human tumor necrosis factor receptor Ⅰ inAspergillusniger[J]. 科学通报(英文版), 2001, 46(11):918-921.
[13] Lichty J J, Malecki J L, Agnew H D,etal.. Comparison of affinity tags for protein purification.[J]. Protein Exp. Purific., 2005, 41(1):98-105.
[14] 苏 鹏, 龚国利. 重组蛋白表达技术的研究进展[J]. 中国酿造, 2016, 35(10):9-12.
[15] 安乃莉, 徐春晓, 姚立红,等. TNF受体(P55)胞外区基因的克隆及其在大肠杆菌中的表达[J]. 病毒学报, 2001, 17(2):127-131.
[16] Fu L, Peng S, Tan D,etal.. Cloning, expression of humansTNFR1 gene and the biological activity of its recombinant protein[J]. China Biotechnol., 2007,27(7):88-93.
[17] Lei F U, Tan D M, Peng S F. Cloning and expression of HumansTNFR1 inE.coliJM109[J]. J. Chin. Phys., 2005,7(7):868-870.
