4例儿童显微镜下多血管炎的肺部CT表现
2018-05-23宋洋
宋洋
显微镜下多血管炎(microscopic polyangiitis,MPA)是一类以系统性坏死性非肉芽肿性血管炎为临床病理特征的血管炎,是抗中性粒细胞胞浆抗体相关性系统性血管炎的一种。原发性MPA是儿童期最多见的血管炎类型,常累及多个系统或器官,可急性起病,表现为急进性肾炎和肺泡出血,也可以隐匿起病,表现为肾、肺、皮肤、消化道和周围神经系统受累,自症状出现到确诊往往达数月甚至数年,影像表现多样且认识不足,相关文献报道较少,易误诊、延误治疗。本文回顾性分析4例MPA的临床和胸部CT影像资料,旨在提高对该病的认识,为临床鉴别诊断提供相关依据。
村料与方法
1.病例资料
搜集2010年7月-2016年10月我院肾内科收治的4例MPA患儿,其中女3例,男1例,年龄3~13岁,中位年龄11岁。1例经肾脏穿刺活检病理确诊 。血清髓过氧化物酶含量用酶联免疫吸附法测定,血清p-ANCA用间接免疫荧光法测定,患儿入院前门诊尿检均有镜下血尿及肾功能异常;就诊时患儿均表现为发热、咳嗽(轻重不一)、尿检异常、中度贫血(血红蛋白降低)等。3例患儿经腹部超声诊断为双肾弥漫性病变,且住院期间4例均出现急性肾衰竭;3例出现眼睑及下肢浮肿,2例血压升高;2例入院前有阵发性腹痛病史,较剧烈,排便后可有缓解,并偶发呕吐,非喷射样;1例患儿曾有下肢骨关节游走性疼痛病史,1例患儿入院期间行超声心动检查示主动脉瓣、二尖瓣、三尖瓣轻度反流。实验室检查结果:WBC(6.0~18.8)×109/L,Hb 40~111 g/L,尿常规RBC(+)~(),ESR 49~103 mm/h,SCr 45~309 μmol/L,BUN 16.6~30.4 mmol/L;4例ANCA均为阳性。MPA确诊后,进行系统治疗,常用皮质激素和环磷酰胺冲击治疗,并予碳酸氢钠、速尿、胰岛素、葡萄糖酸钙静点降低血钾,拮抗高钾及促进细胞外钾向细胞内转移,利尿降压等治疗,治疗两周左右采用胸部CT进行疗效观察。MPA的诊断按美国风湿病学会1990年的诊断标准,每例患者均应有病理活检(肾、肺、皮肤等)或ANCA检测结果,肺部受累是指有呼吸道症状并有影像学证据。
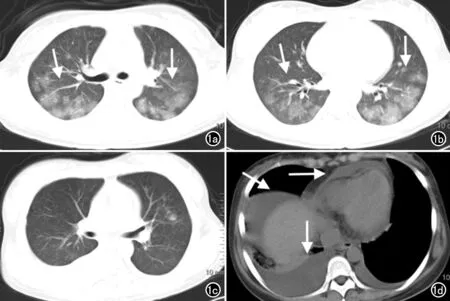
图1 MPA患儿,男,3岁,因发热半月余、尿检异常伴浮肿8天入院,患儿入院后多次行肾功能检查(血Cre 206.3μmol/L,BUN 26.9mmol/L, Ccr 17.67ml/min,伴有低钠血症及高钾血症),提示存在肾衰竭,血Hb61-88g/L,提示贫血,血P-ANCA(±),抗MPO-Ab(±)。B超示双肾弥漫性病变,超声心动图示左心室扩大。a) 轴面CT平扫肺窗示气管分叉层面双肺广泛磨玻璃密度实变影(箭); b) 双肺下叶外带磨玻璃密度实变影尤著(箭); c) 轴面CT平扫肺窗示经治疗后双肺实变影明显吸收好转; d) 轴面CT平扫纵隔窗示双侧胸腔积液、心包积液及腹腔积液(箭)。
2.检查方法
全部患儿均行MSCT检查,采用Siemens Somatom Sensation 16层CT机行横轴面扫描,扫描参数:100 kV,80 mA,层厚4 mm,螺距1.0,卷积核B60s,窗值Baby Spine,对兴趣区行2 mm层厚重组。必要时对原始图像采用多平面重组(multi-planar reformation,MPR)、表面阴影遮盖(surface shaded display,SSD)、容积再现(volume rendering,VR)等技术进行三维重组。由1位初级及1位中级以上影像医师分别使用肺窗和纵隔窗进行阅片,记录如下影像信息,结果不一致时通过协商达成共识:①肺内病变征象。包括磨玻璃影、斑片状实变影、小结节影、小叶间隔增厚、支气管血管束增粗、支气管管壁增厚、牵拉性支气管变形扩张、网格影、空洞形成、蜂窝肺等;②病变分布。病变弥散分布或局灶性分布及确切部位,小结节的部位和分布情况;③胸膜受累情况及是否存在胸腔积液;④纵隔及双腋下、颈部淋巴结是否肿大。
结 果
4例MPA患者肺部CT均显示有不同程度的改变,其中间质性改变3例,2例可见双肺弥漫性磨玻璃密度实变影(图1),复查后可见1例病变部分好转,表现为局限斑片状实变影(图1c),1例呈大片致密实变影。1例可见明显蜂窝肺改变,呈弥漫性网格样变,可见小叶间隔增厚、多发胸膜下线影及双侧胸膜下区多发小气肿(图2a-b)。4例患儿均未见结节样病变。3例患儿病变肺野内可见支气管血管束增粗,管壁增厚,管腔略明显,走行较通畅,未见牵拉性支气管变形扩张。3例伴有双侧胸腔积液(图1d,图2d),2例盆腔亦有积液,1例还可见心包积液(图1d)。1例患儿肺部仅见双肺纹理增重表现,但其双侧胸腔积液较多,盆腔亦有积液。所有患儿均未见明显纵隔及腋下、颈部淋巴结肿大。4例患儿血P-ANCA及抗MPO抗体均为阳性,同时伴有贫血及急性期肾炎实验室指标如ESR、CRP升高、蛋白尿、镜下血尿、血清肌酐和尿素氮水平升高等。1例患儿行肾活检检查,HE染色显示特征性的肾小球节段性坏死、微血栓和新月体形成(图3)。
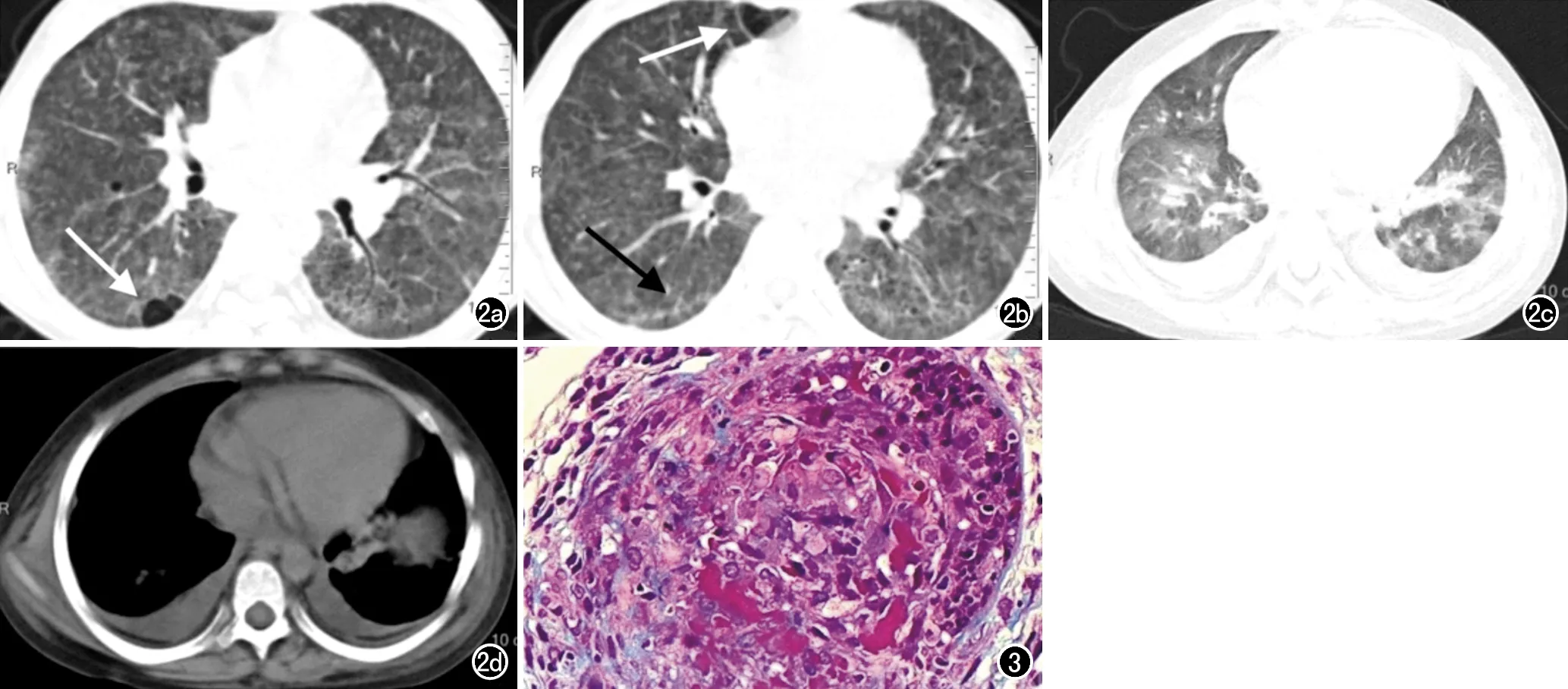
广泛的不规则网状或网结影,右肺下叶可见局限透亮区,考虑间质性肺气肿 (箭);b)右肺下叶可见胸膜下线影(黑箭),右肺中叶可见局限透亮区,考虑间质肺气肿(白箭); c) 复查后胸部CT平扫肺窗示双肺透过度不均匀,可见散在磨玻璃密度影; d) 复查后胸部CT平扫纵隔窗示双侧胸腔积液。 图3 MPA患儿(经病理确诊),男,3岁,肾活检可见特征性肾小球节段性坏死、微血栓和新月体形成(×400,HE) 。
图2 MPA患儿,女,12岁,因面色苍白2个月,尿检异常伴浮肿3天入院,患儿血P-ANCA及抗MPO抗体(+),无高丙球血症及低补体血症,ANA、ENA(-)。a) 轴面胸部CT平扫肺窗示双肺弥漫多发磨玻璃密度渗出性病变,可见对称的
讨 论
1.MPA的病理特征及临床表现
MPA最先由Davson等报道,美国风湿病学院1990年血管炎分类标准中,把MPA包括在结节性多动脉炎(polyarteritisnodosa,PAN)中。1994年Chapel Hill会议将MPA从PAN中独立出来,2012年Chapel Hill会议上再次规范了血管炎分类标准,将血管炎分为大、中、小血管血管炎,小血管血管炎包括抗中性粒细胞胞浆抗体(Anti-neutrophil cytoplasmic antibodies,ANCA)相关性小血管炎、免疫复合物小血管炎、变异性血管炎、单器官性血管炎、与系统性疾病相关的血管炎、与可能的病因相关的血管炎。其中ANCA相关性小血管炎主要包括MPA、肉芽肿性多血管炎以及嗜酸粒细胞性肉芽肿性血管炎。儿童期发病率最高的小血管血管炎就是MPA。
MPA是一种系统性、非肉芽肿性、免疫组织学检查少或无免疫复合物沉积的小血管炎。该病主要依靠临床和组织病理学检查确诊,病程早期多累及肾和肺。最常见的肺部病理结果是中性粒细胞毛细血管炎和急、慢性肺泡出血[1-2]。
MPA临床表现多样,可有发热、乏力、肌肉关节疼痛、纳差、消瘦等全身症状,亦有肾、心、肺、消化、神经、皮肤、眼等多系统受累表现。其中肺部受累(>90%)可有咯血、呼吸困难、贫血等症状。肾脏受累(>90%)主要表现为血尿、蛋白尿及不同程度肾功能不全。消化系统受累(>50%)可表现为腹痛、腹泻、腹胀、肠梗阻等。本组4例患儿肺部均受累,并经尿检及病理活检[P-ANCA(±),抗MPO-Ab(±)]提示肾脏亦全部受累。4例患儿中2例肠道受累,表现为明显腹痛,偶发呕吐。4例均未见明显皮肤及神经系统受累表现。本病容易继发肺感染,肺感染与MPA肺部受累相叠加可协同损害呼吸功能。本组有2例患儿最终死亡,直接死于弥漫性肺泡出血并呼吸衰竭者较少,故考虑死因可能为肺部感染,提示肺感染是影响MPA预后的主要因素。
2.MPA的CT表现
欧洲血管炎研究组已建议将肺部CT检查作为系统性血管炎的筛查项目之一,因为该病肺部影像表现多样,但无明显特异性,仅依靠胸部X线平片很难确诊。肺泡的毛细血管炎导致的弥漫性肺泡出血是MPA肺部受累最常见的表现,发生在29%以上的患者中[3],急性期CT表现为肺纹理增重、模糊,双侧弥漫性的肺泡渗出性病变,弥漫性肺泡出血等,肺泡完全充填时较为致密,呈片状或云片状,肺泡部分充填时呈磨玻璃密度影,较为稀薄,累及多个肺叶;本组3例患儿有此影像表现,有1例发生于发病初期,有1例治疗一段时间后复查发现此征象,有1例患儿病程迁延较久,呈慢性持续性或反复发作的弥漫性肺泡出血,可引起阻塞性肺疾病,伴随有肺气肿[4-5]和肺间质纤维化[5]。MPA患者肺间质纤维化的发生率约为36%[5],表现为对称的、广泛的不规则网状或网结影,以中、下叶和肺外带多见,可见小叶间隔增厚及支气管管壁增厚等。本组有3例表现出肺间质纤维化的特征。相关文献报道,胸膜炎伴或不伴积液不多见[3],但本组4例患儿均出现了胸膜增厚和/或胸腔积液。部分患者肺部可以没有任何改变,此时可通过肾脏穿刺明确诊断,本组中有1例肺部未见明显病变影,仅见双肺纹理粗重。分析4例患儿的CT表现,发现MPA的CT表现没有明显特异性,需与多种呼吸道疾病进行鉴别,并结合相关临床特点、实验室检查及病理学检查结果进行综合分析。
3.鉴别诊断
与特发性肺间质纤维化的鉴别:两者病理基础不同,特发性肺间质纤维化早期肺泡间隔即有不同程度的细胞浸润,肺泡腔可见巨噬细胞,继之纤维成分增多,肺泡结构破坏,可扩大融合成囊状。相关学者认为MPA肺损害以肺间质纤维化开始,即肺间质改变可能是MPA早期重要的临床表现,可先于肺出血长期隐匿存在,病程后期出现咯血应视为疾病恶化的表现[1]。
与特发性肺含铁血黄素沉着症的鉴别:该病以弥散性肺泡内出血及继发缺铁性贫血为特征,急性出血期表现为肺内磨玻璃影及大片云絮状实变影,双侧多发,多见于肺门及中下肺野;出血静止期表现为双肺纹理增重,模糊毛糙;慢性反复发作期表现为肺透过度减低,肺野内广泛分布的境界模糊的细网状阴影,小叶间隔及肺泡壁增厚;病程迁延反复者表现为双肺弥漫分布的粟粒样病灶或粗网粒结构,间质纤维增生,严重者可发展为弥漫性肺间质纤维化。痰及胃液细胞学分析查找含铁血黄素细胞能协助诊断。
与过敏性肺炎的鉴别:急性期过敏性肺炎病理特点为肺泡水肿及肺间质炎症,CT表现特点为弥漫性磨玻璃密度增高影及马赛克灌注,慢性期表现为纤维化增生,伴有网格样蜂窝肺改变。诊断时应全面了解临床病史及相关实验室检查,排除遗传及过敏源接触史。
综上所述,MPA的CT表现无明显特异性,对于临床上长期不规则发热、血尿、蛋白尿、气喘、不明原因咯血、贫血的儿童,肺部影像学表现为多发斑片影或弥漫性间质改变,以及血清p-ANCA和抗MPO检测呈阳性时,即可考虑MPA的可能。
参考文献:
[1] Lohrmann C,Uhl M,Kotter E,et al.Pulmonary manifestations of wegener granulomatosis:CT findings in 57 patients and a review of the literature[J].Eur J Radiol,2005,53(3):471-477.
[2] Tzelepis GE,Kokosi M,Tzioufas A,et al.Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis[J].Eur Respir J,2010,36(1):116-121.
[3] Klemmer PJ,Chalermskulrat W,Reif MS,et al.Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis[J].Am J Kidney Dis,2003,42(6):1149-1153.
[4] Slot MC,Tervaert JW,Boomsma MM,et al.Positive classic antineutrophil cytoplasmic antibody (C-ANCA)titer at switch to azathioprine therapy associated with relapse in proteinase 3-related vasculitis[J].Arthritis Rheum,2004,51(2):269-273.
[5] Reinhold-Keller E,Fink CO,Herlyn K,et al.High rate of renal relapse in 71 patients with Wegener's granulomatosis under maintenance of remission with low-dose methotrexate[J].Arthritis Rheum,2002,47(3):326-332.
