G蛋白偶联雌激素受体1对血管紧张素Ⅱ诱导心肌细胞肥大的影响及其机制
2018-05-21裴慧王立启王磊王维毕秀萍才晓君苏国海赵卓
裴慧,王立启,王磊,王维,毕秀萍,才晓君,苏国海,赵卓
内源性雌激素对女性绝经前心血管内环境的稳定起重要的调节作用[1],能延缓高血压、心力衰竭和冠状动脉疾病的发展。绝经后雌激素替代疗法对心血管疾病具有保护作用[2,3]。最初研究证实,雌激素主要通过经典雌激素受体ERα和ERβ来调控基因的转录进而保护心血管系统[4,5]。随着研究的深入,G蛋白偶联受体30(GPR30)作为雌激素受体的功能被发现[6],国际药理学联合会(IUPHAR)也将GPR30或G蛋白偶联雌激素受体1(GPER1)分类为膜雌激素受体。
GPER1参与雌激素介导的基因转录和信号通路的调节[7]。GPER1主要通过快速非基因组机制参与雌激素信号的传导,如细胞应答效应中的钙动员、激酶激活及一氧化氮产生等[8]。近年来,特异性GPER1激活剂G1(简称G1)[9]和抑制剂G15(简称G15)的合成,为其药理学和生理学的研究开辟了新途径。研究发现,G1作用于内皮细胞、血管平滑肌细胞和肾素-血管紧张素系统,产生舒张血管[10]、降低血压[11]、抗炎[12]、防止心肌再灌注损伤[13]、缓解肺动脉高压[14]等心血管保护作用。
虽然GPER1具有潜在的心血管保护作用,但其在缓解心肌细胞肥大中的作用机制仍未阐明。本研究通过串联质谱标签(TMT)蛋白质谱技术和生物信息学分析进行大数据分析,通过蛋白免疫印迹法(Western-blot)和荧光定量逆转录聚合酶链式反应(qRT-PCR)技术探究其可能的调控机制。
1 材料与方法
材料与试剂: G1、G15(Cayman Chemical,美国),DMEM高糖培养基(Hyclone,美国),胎牛血清(Gibico,美国)、马血清(BI,以色列),血管紧张素Ⅱ(AngⅡ)、抗GPER1抗体(Abcam,美国),抗丝氨酸/苏氨酸激酶(AKT)抗体、抗p-AKT抗体、抗细胞外调节蛋白激酶(ERK)抗体、抗p-ERK抗体、抗LC3抗体和抗GAPDH抗体(CST,美国),二抗HRP(Proteintech,中国),Alexa Fluor 488抗体(Life Technologies,美国),4',6-二脒基-2-苯基吲哚(DIPI,Thermo Fisher Scientific,美国),qRT-PCR试剂盒(TAKARA,日本)和流式试剂盒(BD,美国)。
细胞培养:2~3日龄的新生Wistar乳鼠50只(购自山东大学动物实验中心)表皮消毒后,摘取心脏并剪碎成2~3 mm3的组织块,胰蛋白酶(0.1%)重悬置于磁力搅拌器消化10 min。5%胎牛血清的DMEM培养基终止消化,800 g离心10 min后收集细胞。DMEM高糖培养基(含有6%马血清、8%新生牛血清和1%青霉素/链霉素),培养箱中培养细胞24 h。
模型建立和分组:心肌细胞血清饥饿后,AngⅡ(0 、50 、100 和250 nmol/L)诱导心肌细胞肥大。检测信号通路检测实验分为6组:空白对照组、AngⅡ 组、AngⅡ +G1组、AngⅡ +G1+G15组、AngⅡ+G1+ERK抑制剂 (U0126)组和AngⅡ+G1+AKT抑制剂 (MK2206)组。空白对照组不给药;AngⅡ组给予AngⅡ(100 nmol/L)处理;AngⅡ+G1组给予AngⅡ(100 nmol/L)和G1(100 nmol/L)处理;AngⅡ+G1+G15组给予AngⅡ(100 nmol/L)、G1(100 nmol/L)和G15(100 nmol/L)处理;AngⅡ +G1+U0126组 给 予 AngⅡ(100 nmol/L)、G1(100 nmol/L) 和 U0126(100 nmol/L) 处 理;AngⅡ+G1+MK2206组给予AngⅡ(100 nmol/L)、G1(100 nmol/L)和 MK2206(5 μmol/L)处理。各组n=3。
蛋白质谱分析:检测空白对照组、AngⅡ(100 nmol/L)组和AngⅡ+G1(100 nmol/L)组的蛋白表达差异。用软件Mascot2.5和Proteome Discoverer2.1,筛选出差异倍数>1.2,P<0.05的差异蛋白分子。
细胞免疫荧光染色:心肌细胞4%多聚甲醛固定20 min,0.5%Triton X-100细胞膜通透剂室温通透20 min,血清封闭。GPER1抗体1:300稀释4℃孵育过夜,PBST(PBS+Tween-20洗液,pH=7.4)浸洗后,荧光二抗避光孵育1 h,DAPI避光孵育10 min,镜下观察采集图像。
Western-blot: 6 孔板加入 100 µl RIPA 裂解液和1%PMSF蛋白酶抑制剂裂解细胞,超声粉碎,4℃12 000 g离心,取上清蛋白定量。25 µl 5 X loading buffer 混匀后100℃水浴,-80℃保存。SDS-聚丙烯酰胺凝胶电泳,90 V恒压电泳,250 mA恒流转膜,5%脱脂奶粉封闭,洗膜,抗体1:1 000稀释4℃孵育过夜。TBST洗液漂洗后,二抗1:1 0000孵育1 h,漂洗后化学发光法显色。
qRT-PCR检测: Trizol法提取总RNA后,按Takara试剂盒说明书要求行cDNA逆转录。所有引物购自Takara公司,引物序列:心房钠尿肽(ANP)上 游 引 物5'-CGTATACAGTGCGGTGTCCA-3',下 游 引 物 5'-GGTTGACTTCCCCAGTCCAG-3';B型 利 钠 肽(BNP) 上 游 引 物5'-AGCTGCTGGAGCTGATAAGAG-3', 下 游 引 物5'-CTGCCCAAAGCAGCTTGAAC-3';GPER1 上 游 引物5'-CCATCATCGGCCTGTGCTAT-3',下游引物5'-GAAGACAAGGACCACTGCGA-3';GAPDH 上游引物5'-AAGAAGGTGGTGAAGCAGGC-3′,下游引物5'-TCCACCACCCTGTTGCTGTA-3'。
统计学方法:数据采用SPSS 20.0和GraphPad Prism 5软件进行分析。计量资料以±s表示,组间比较用单因素方差分析,以P<0.05为差异有统计学意义。
2 结果
2.1 血管紧张素Ⅱ诱导心肌细胞肥大
Ang Ⅱ(0、50、100 和 250 nmol/L)诱导心肌细胞24 h。各组心肌细胞表面积分别为(1 343.6±202.9)μm2,(1 657.3±168.6)μm2,(2 603.1±381.7)μm2, (3 350.0±413.3)μm2。qRT-PCR检测结果,与空白对照组相比,AngⅡ(100 nmol/L) 组的心肌细胞表面积明显增大(P<0.01)。
2.2 心肌细胞中存在G蛋白偶联雌激素受体1的表达(图 1)
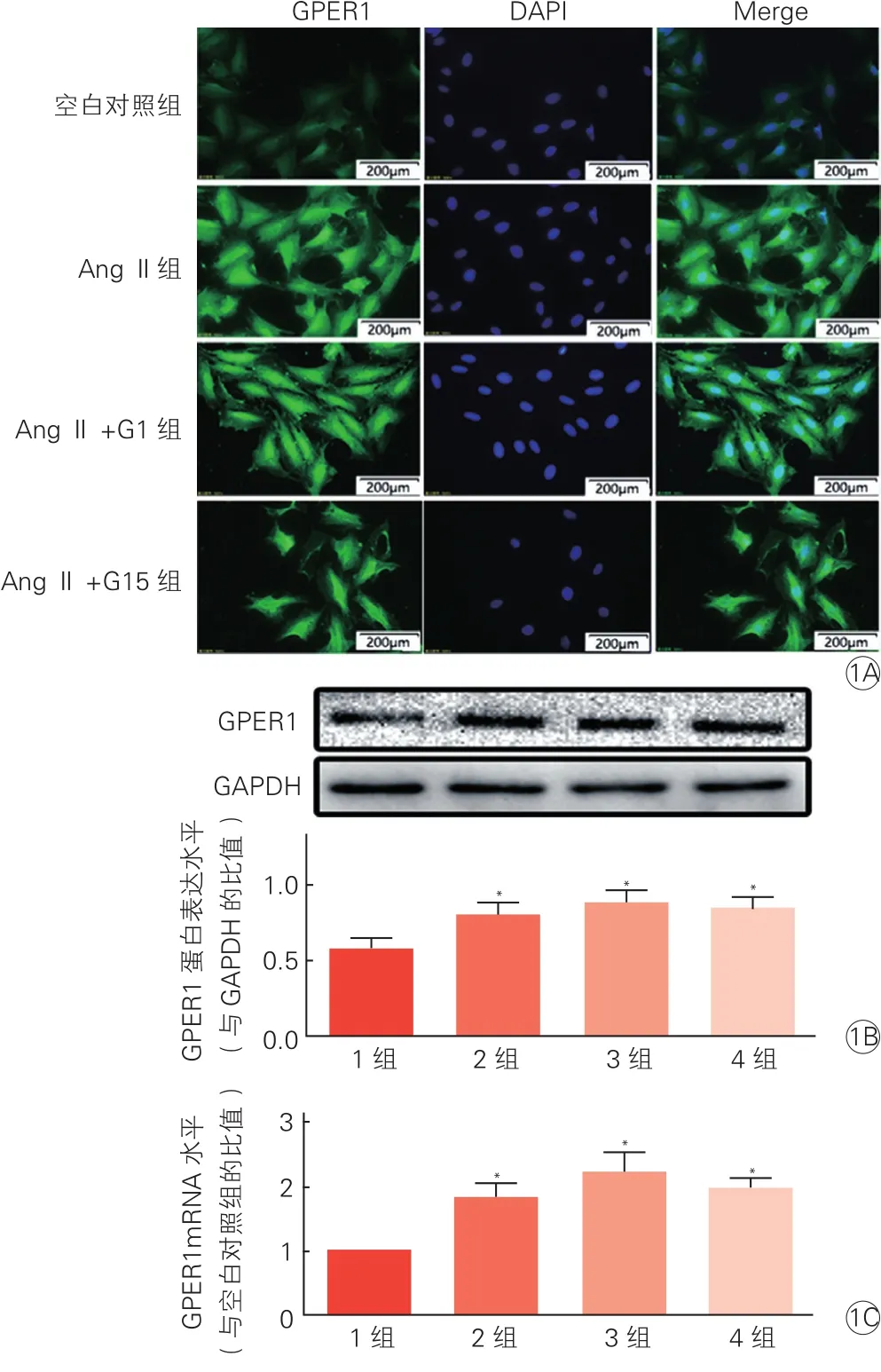
图1 细胞免疫荧光染色、蛋白免疫印迹法、荧光定量逆转录聚合酶链式反应监测心肌细胞G蛋白偶联雌激素受体1的表达
心肌细胞特异性免疫荧光(抗GPER1,绿光)染色结果显示(图1A),AngⅡ组、AngⅡ+G1组和AngⅡ+G15组肥大心肌细胞GPER1蛋白表达均较空白对照组明显增加。Western-blot结果显示(图1B),AngⅡ组、AngⅡ+G1组和AngⅡ+G15组肥大心肌细胞GPER1蛋白表达水平均较空白对照组略有升高(P均<0.05),AngⅡ+G1组和AngⅡ+G15组对GPER1蛋白表达无差异(P>0.05)。qRT-PCR结果显示(图1C),AngⅡ组、AngⅡ+G1组和AngⅡ+G15组肥大心肌细胞GPER1 mRNA水平均较空白对照组均明显升高(P均<0.05)。
2.3 激活G蛋白偶联雌激素受体1可以降低心肌细胞心房钠尿肽和B型利钠肽信使核糖核酸水平(图2)
GPER1激活剂G1(10、100和1 000 nmol/L)和AngⅡ(100 nmol/L)共同作用于心肌细胞。qRT-PCR结果显示,与空白对照组相比,AngⅡ(100 nmol/L)组和AngⅡ+G1(10、100和1 000 nmol/L)组肥大心肌细胞ANP和BNP mRNA水平均明显升高(P<0.01)。随着G1浓度的升高,ANP和BNP mRNA水平基本呈梯度降低,尤其是AngⅡ+G1(100、1 000 nmol/L)组与AngⅡ(100 nmol/L)组相比ANP和BNP mRNA水平均明显降低(P均<0.05)。
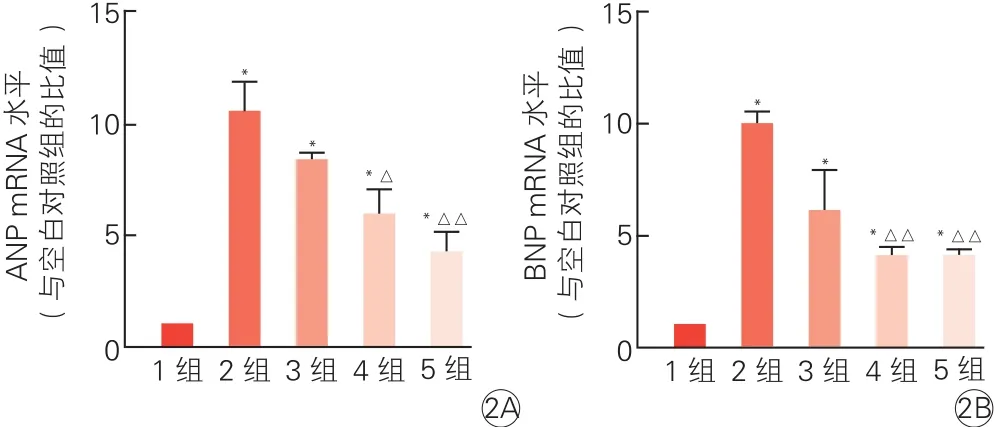
图2 荧光定量逆转录聚合酶链式反应检测浓度梯度G蛋白偶联雌激素受体1激活剂对心肌细胞心房钠尿肽(2A) 和B型利钠肽信使核糖核酸(2B)的影响
2.4 各组乳鼠心肌细胞蛋白质质谱分析
蛋白质谱分析显示:空白对照组与AngⅡ组组间差异蛋白52种;AngⅡ组与AngⅡ+G1组组间差异蛋白36种。两组间差异蛋白交叉存在的5种关键基因是Fam120b、RGD1562310、Ctrb1、Rap1gap和Pag1。与空白对照组相比,AngⅡ组的Rap1gap蛋白高表达(比值=1.348 98,P=0.033 99),而与AngⅡ组相比,AngⅡ+G 1组的Rap1gap蛋白表达又相对低表达(比值=0.743 79,P=0.017 02)。
2.5 激活G蛋白偶联雌激素受体1参与调控心肌细胞的信号通路(图3)
Western-blot结果显示,与空白对照组或AngⅡ组相比,AngⅡ+G1组p-AKT和p-ERK蛋白表达均有显著升高(P<0.01)。AngⅡ+G1+G15组与AngⅡ+G1组相比,p-AKT和p-ERK蛋白表达降低(P均<0.05)。AngⅡ+G1+U0126组与 AngⅡ +G1组相比,p-ERK蛋白表达水平降低(P均<0.01),而p-AKT蛋白表达水平无变化(P>0.05);AngⅡ+G1+MK2206组与AngⅡ+G1组相比,其p-AKT和p-ERK蛋白表达水平均有明显降低(P均<0.01)。
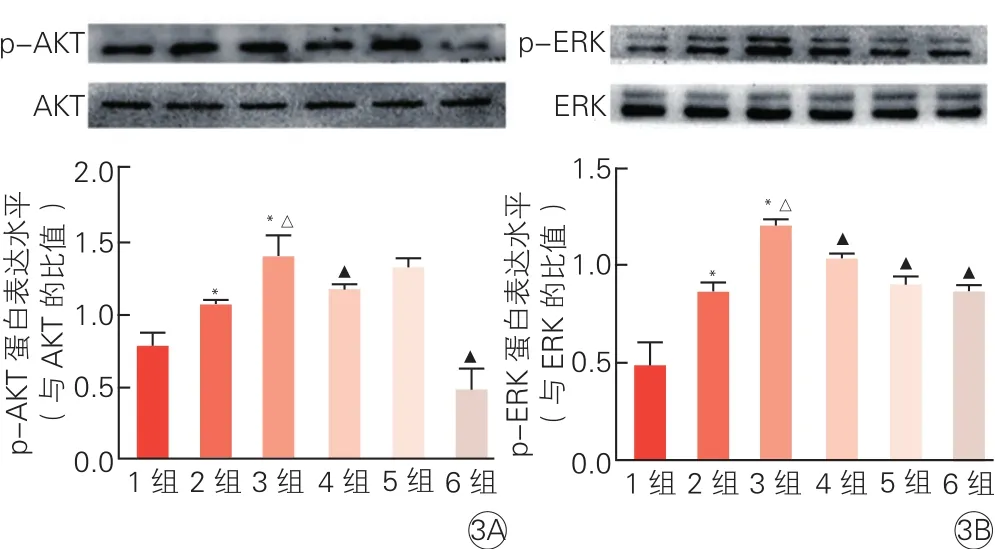
图3 蛋白免疫印迹法检测6组心肌细胞丝氨酸/苏氨酸激酶(3A)和细胞外调节蛋白激酶蛋白磷酸化(3B)水平的影响
2.6 G蛋白偶联雌激素受体1参与缓解心肌细胞肥大可能主要与丝氨酸/苏氨酸激酶信号通路有关(图4)
qRT-PCR结果显示,与空白对照组比,AngⅡ组ANP和BNP mRNA水平均有显著升高(P<0.01)。与AngⅡ组比,AngⅡ+G1组ANP和BNP mRNA水平均有明显降低(P<0.01)。与 AngⅡ组或AngⅡ+G1组相比,AngⅡ+G1+MK2206组ANP和BNP mRNA水平均有明显升高(P均<0.01)。
2.7 激活G蛋白偶联雌激素受体1对心肌细胞凋亡和自噬的影响(图5)
流式细胞技术检测结果显示,空白对照组、AngⅡ组和AngⅡ+G1组的凋亡水平差异无统计学意义(P>0.05)。Western-blot结果显示,与空白对照组相比,AngⅡ组LC3II/LC3I增大(P<0.01);与AngⅡ组相比,AngⅡ+G1组LC3II/LC3I减小(P<0.01)。与AngⅡ+G1组相比,AngⅡ+G1+G15组心肌细胞LC3II/LC3I比值升高(P<0.01)。
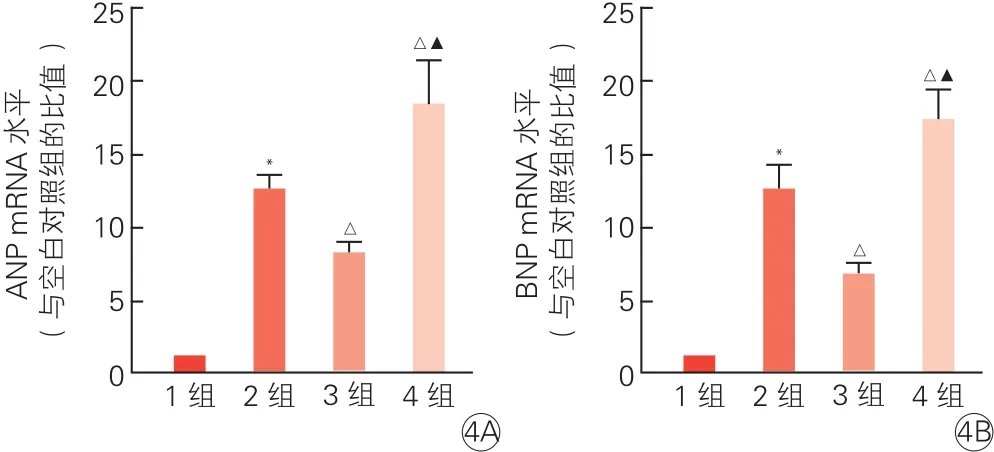
图4 荧光定量逆转录聚合酶链式反应检测丝氨酸/苏氨酸激酶抑制剂对心肌细胞心房钠尿肽(4A)和B型利钠肽信使核糖核酸(4B)水平的影响
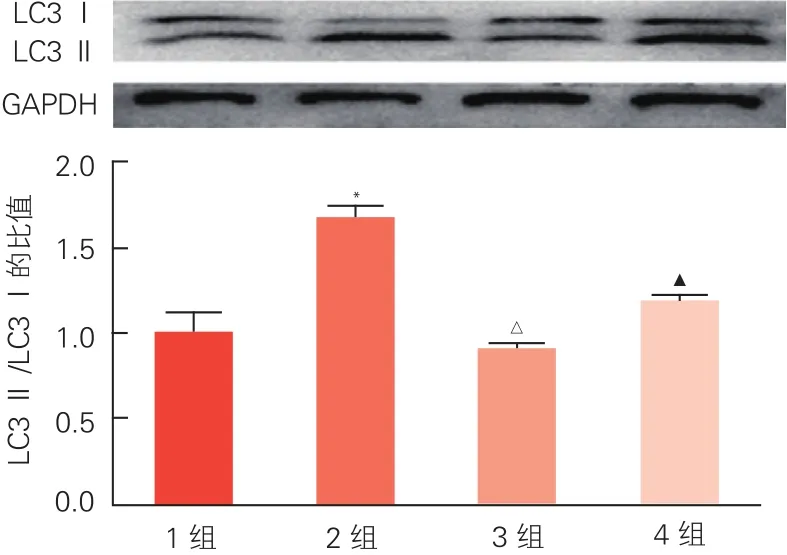
图5 蛋白免疫印迹检测激活G蛋白偶联雌激素受体1对心肌细胞胞浆型自噬标记轻链3和膜型自噬标记轻链3表达的影响
3 讨论
女性健康指南(WHI)随机控制试验和心脏雌激素重施(HERS)试验均未得出雌激素对心血管系统保护作用的支持性结论,并且绝经期女性外源性雌激素替代治疗可能增加女性生殖系统肿瘤[15],限制了临床应用雌激素来保护心肌的相关研究的开展。新型膜雌激素受体GPER1的发现为雌激素抗心血管疾病的研究带来了新的希望。本研究主要探讨激活GPER1快速信号转导途径对心肌细胞的影响。本研究通过免疫荧光染色,Western-blot和qRT-PCR技术发现,大鼠原代心肌细胞存在GPER1的表达,这与De Francesco等[16]的研究结果是相互印证的。
缓解心肌细胞肥大是防止心脏肥厚进一步恶化的关键。ANP和BNP是重要的心脏神经内分泌激素,与心脏压力负荷和容量负荷的增加有关,其mRNA水平是衡量心肌肥厚和心力衰竭的重要指标[17]。激活的GPER1能有效缓解心肌细胞肥大,该作用可被G15和MK2206阻断。这就为雌激素替代疗法提供了新的可能。单纯激活GPER1既可以缓解细胞肥大,又相对之前的雌激素替代疗法更加安全。
生物信息学分析结果显示Rap1gap蛋白分子下游的B-Raf/Raf-1-MEK-ERK信号通路、MAPK信号通路和PI3K-AKT信号通路参与GPER1介导的心肌细胞调控的关键信号通路。ERK信号通路与细胞的核转录作用有关,而AKT信号通路可能与心肌细胞的生长、肥大和存活相关。本研究Western-blot结果显示,AKT和ERK参与GPER1介导的抗心肌细胞肥大作用,且MK2206(AKT特异性抑制剂)可同时抑制p-AKT和p-ERK蛋白表达。这就说明,PI3K-AKT信号通路和MEK-ERK信号通路可能存在交叉,AKT可能参与调控ERK的表达。
哺乳动物的雷帕霉素靶(mTOR)作为AKT下游重要蛋白分子,是一种非典型AKT,可以影响基因转录与蛋白质翻译,参与调控细胞生长、增殖等过程,与蛋白质合成、免疫、细胞运动及代谢、细胞凋亡及自噬等均有联系[18]。相关实验证实,雷帕霉素能抑制心肌细胞肥大,肥大过程中蛋白质合成的增加与AKT-mTOR信号通路的激活有关,雷帕霉素和PI3K抑制剂均能抑制肥大过程中蛋白质的合成[19]。细胞凋亡和自噬作为细胞维持内环境稳态的重要机制,都有可能参与心肌细胞肥大的调控。但本研究证实,GPER1介导的抗心肌细胞肥大作用可能与细胞凋亡无关,而与细胞自噬相关。GPER1受体可能通过PI3K-AKT-mTOR信号通路介导细胞自噬发挥抗心肌细胞肥大作用。然而,自噬与mTOR通路之间的关系非常复杂,而且GPER1的激活mTOR的具体机制,以及TORC1和mTORC2的相互调节作用仍需要进一步研究揭示。
我们目前的研究尚处于起始阶段,对GPER1生物功能的理解还不够深入,随着研究的深入,GPER1的潜在致癌作用(特别是妇科癌症,如卵巢癌[20]和乳腺癌[21])和心血管保护功能将逐渐被揭示。
参考文献
[1] Kearney PM, Whelton M, Reynolds K, et al. Global burden of hypertension: analysis of worldwide data[J]. Lancet, 2005, 365(9455):217-223. DOI: 10. 1016/S0140-6736(05)17741-1.
[2] 张冬, 窦克非. 绝经期前女性冠心病发病机制研究进展[J]. 中国循环杂志, 2012, 27(5): 397-398. DOI: 10. 3969/j. issn. 1000-3614.2012. 05. 023.
[3] Baber RJ, Panay N, Fenton A, et al. 2016 IMS Recommendations on women's midlife health and menopause hormone therapy[J].Climacteric, 2016, 19(2): 109-150. DOI: 10. 3109/13697137. 2015.1129166.
[4] Arias-loza PA, Jazbutyte V, Fritzemeier KH, et al. Functional effects and molecular mechanisms of subtype-selective ERalpha and ERbeta agonists in the cardiovascular system[J]. Ernst Schering Found Symp Proc, 2006, 16(1): 87-106. DOI: 10. 1007/2789. 2006. 018.
[5] Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets[J]. Physiol Rev, 2007, 87(3):905-931. DOI: 10. 1152/physrev. 00026. 2006.
[6] Revankar CM, Cimino DF, Sklar LA, et al. A transmembrane intracellular estrogen receptor mediates rapid cell signaling[J].Science, 2005, 307(5715): 1625-1630. DOI: 10. 1126/science.1106943.
[7] Meyer MR, Haas E, Prossnitz ER, et al. Non-genomic regulation of vascular cell function and growth by estrogen[J]. Mol Cell Endocrinol,2009, 308(1-2): 9-16. DOI: 10. 1016/j. mce. 2009. 03. 009.
[8] Prossnitz ER, Arterburn JB, Smith HO, et al. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30[J].Annu Rev Physiol, 2008, 70(1): 165-190. DOI: 10. 1146/annurev.physiol. 70. 113006. 100518.
[9] Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30[J]. Nat Chem Biol, 2006, 2(4): 207-212. DOI: 10. 1038/nchembio775.
[10] Knowlton AA, Lee AR. Estrogen and the cardiovascular system[J].Pharmacol Ther, 2012, 135(1): 54-70. DOI: 10. 1016/j. pharmthera.2012. 03. 007.
[11] Seok YM, Jang EJ, Reiser O, et al. 17beta-Estradiol induces vasorelaxation in a G-protein-coupled receptor 30-independent manner[J]. Naunyn Schmiedebergs Arch Pharmacol, 2012, 385(9):945-948. DOI: 10. 1007/s00210-012-0770-y.
[12] Chakrabarti S, Davidge ST. G-protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation[J]. PloS One, 2012, 7(12):e52357. DOI: 10. 1371/journal. pone. 0052357.
[13] Lee TM, Lin SZ, Chang NC. Both GPER and membrane oestrogen receptor-alpha activation protect ventricular remodelling in 17beta oestradiol-treated ovariectomized infarcted rats[J]. J Cell Mol Med,2014, 18(12): 2454-2465. DOI: 10. 1111/jcmm. 12430.
[14] 郑泉, 邓华君. 雌激素及其代谢产物与肺动脉高压的相关性研究[J]. 中国循环杂志, 2015, 30(2): 199-200. DOI: 10. 3969/j. issn.1000-3614. 2015. 02. 027.
[15] Pardini D. Hormone replacement therapy in menopause[J]. Arq Bras Endocrinol Metabol, 2014, 58(2): 172-181. DOI: 10. 1590/0004-2730000003044.
[16] De Francesco EM, Angelone T, Pasqua T, et al. GPER mediates cardiotropic effects in spontaneously hypertensive rat hearts [J]. PloS One, 2013, 8(8): e69322. DOI: 10. 1371/journal. pone. 0069322.
[17] Maisel A. B-type natriuretic peptide levels: diagnostic and therapeutic potential[J]. Cardiovasc Toxicol, 2001, 1(2): 159-164. DOI: 10. 1385/CT: 1: 2: 159.
[18] Lee CH, Inoki K, Guan KL. mTOR pathway as a target in tissue hypertrophy[J]. Annu Rev Pharmacol Toxicol, 2007, 47(1): 443-467.DOI: 10. 1146/annurev. pharmtox. 47. 120505. 105359.
[19] Hafizi S, Wang X, Chester AH, et al. AngⅡ activates effectors of mTOR via PI3-K signaling in human coronary smooth muscle cells[J].Am J Physiol Heart Circ Physiol, 2004, 287(3): H1232-1238. DOI: 10.1152/ajpheart. 00040. 2004.
[20] Ho SM. Estrogen, progesterone and epithelial ovarian cancer[J].Reprod Biol Endocrinol, 2003, 1(1): 73. DOI: 10. 1186/1477-7827-1-73.
[21] Jiang QF, WU TT, Yang JY, et al. 17beta-estradiol promotes the invasion and migration of nuclear estrogen receptor-negative breast cancer cells through cross-talk between GPER1 and CXCR1[J]. J Steroid Biochem Mol Biol, 2013, 138(292): 314-324. DOI:10. 1016/j.jsbmb. 2013. 07. 011.
