脂质沉积性肌病18例临床分析
2018-05-21蔡少鹏
田 冉, 蔡少鹏
自EngeL AG 1973年首次报道脂质沉积性肌病( lipid storage myopathy,LSM)以来,有关本病的散发报道日渐增多。该病的临床表现复杂多样,与重症肌无力、多发性肌炎、肢带型肌营养不良、慢性吉兰-巴雷综合征等鉴别困难[1,2]。本文通过对18例经病理证实的脂质沉积性肌病的临床表现、病理特点及常见的误诊诊断作一总结,以增加对此病的认识。
1 临床资料
1.1 一般资料 2006~2011年我院肌萎缩科收治的经病理确诊的18例脂质沉积性肌病患者。男性14例,女性4例,发病年龄12~67岁。平均起病年龄为(31±14)岁。病程0.25~9 y,平均(4.5±2.9)y。其中4例有家族史,3例同胞姐弟均为此病,1例患者的女儿确诊为此病。首次误诊为多发性肌炎者10例、进行性肌营养不良者4例、重症肌无力者1例。
1.2 临床特点 (1)起病形式:除2例为亚急性起病外,其余16例均为渐进性起病。(2)诱因:3例为感冒,2例为劳累,其余13例均无明显诱因。(3)首发症状:四肢或双下肢无力者17例(占94%);咀嚼、吞咽困难者1例(占6%)。(4)主要临床表现:18例患者均表现为易疲劳,休息后缓解(占100%),伴有颈肌无力者7例(占38.9%),声音嘶哑、饮食呛咳、咀嚼、吞咽困难者7例(占38.9%),肌肉酸痛者11例(占61%),心慌、活动后气短2例(占11%);伴咳嗽者2例(占6%),恶心、烧心、反酸者2例(占6%),伴有双下肢水肿者1例(占5%)。(5)体征:近端肌力4~5级12例,3~4级6例;受累肌群萎缩者6例(占33.3%,均以肢体近端肌肉萎缩为主);肌肉压痛者10例(占55.6%);腱反射减弱者10例(占55.6%);Gower征阳性者1例(5% )。所有患者疲劳试验均阳性。
1.3 实验室检查 本组中所有患者均行心肌酶谱检查,CK增高者12例(66.7%,232~6307 U/L)。肌酸激酶总活性<1000 U/L 6例,1000~2000 U/L1例,>2000U/L 1例。肝功能异常者10例,以ALT或AST增高为主。18例患者均行肌电图检查,结果显示肌电图均异常。其中单纯肌源性损害者15例(83.3% ),与神经源性损害共存者3例(16.7%),未发现单纯神经源性损害。重复频率电刺激1例,为低频无衰减,高频无递增。所有患者行心电图检查,其中4例患者心电图异常,心律不齐者2例,窦性心动过缓者1例,1例为频发房性期前收缩,伴室内差异性传导。胸片检查结果:1例胸片为左中肺野钙化点,1例肺CT为两肺下叶少许间质性变。
1.4 肌肉活检 所有患者均行肌肉活检检查。由病理医师和临床医师根据患者的肌肉萎缩情况、肌电图、肌酶、肌力等指标确定取材部位。每例患者染色种类为HE染色、Gomori改良染色、PAS染色、脂肪染色(SBB)、NADH染色、ATP酶染色(pH 4.3、pH 9.4)、茜草红(CA)血管染色等。组织病理结果:18例患者SSB染色均为阳性(100%),肌纤维内脂滴显著增多。伴神经纤维轻度损伤者3例(16.7%),CA轻度阳性者3例(16.7%)。HE染色见肌细胞大小形态无明显改变,部分肌纤维有轻度空泡样改变。ATP染色可见I,II型纤维镶嵌排列,部分有成簇现象。糖原含量无明显增多,肌纤维间及血管旁未找到明显炎细胞浸润,小血管未见增生,血管壁薄厚均匀。见图1~图4。
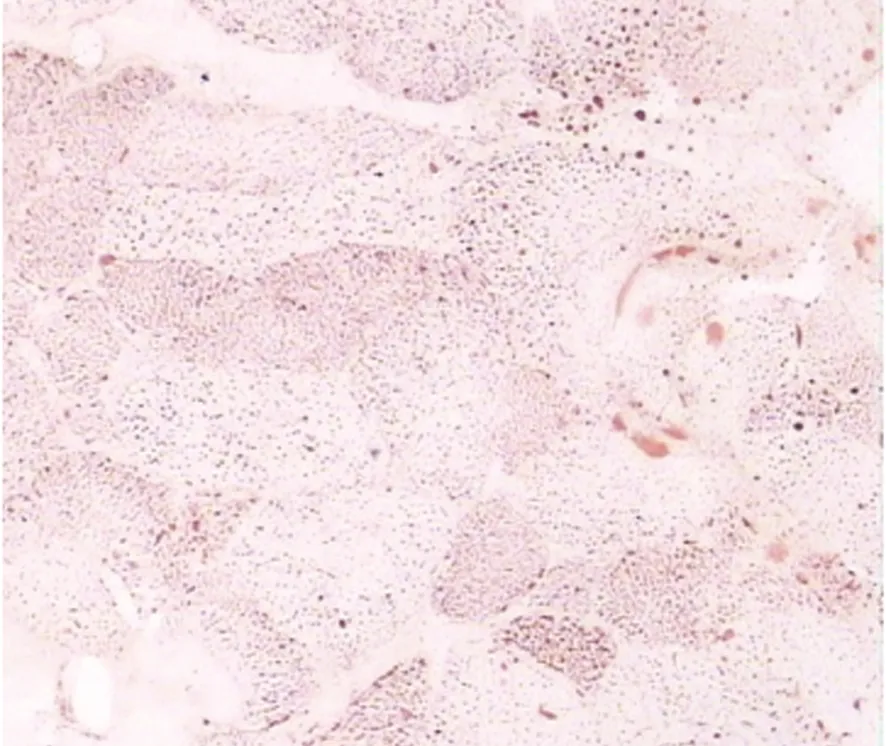
图1 大量肌纤维内脂滴显著增多(ORO 染色×200)
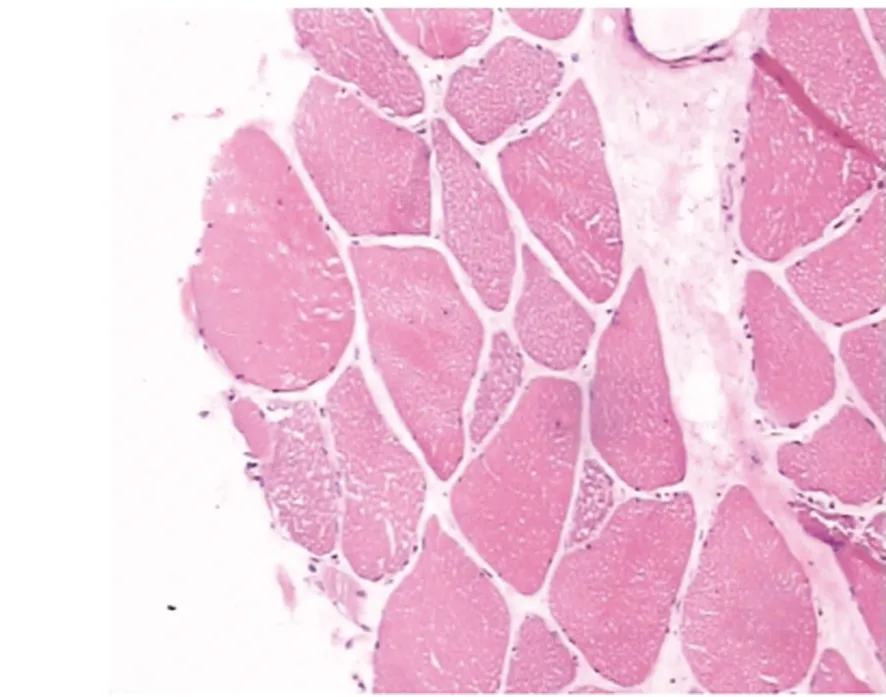
图2 部分呈细小空泡样改变(HE 染色×200)

图3 Ⅰ型肌纤维呈细小空泡样改变(NADH 染色×200)

图4 Ⅰ型、Ⅱ型肌纤维呈镶嵌排列(ATP 染色× 200)
2 讨 论
脂质沉积性肌病(lipid storage myopathy,LSM)是由于脂质代谢途径中,由于缺乏肉毒碱或(和)一些酶,直接或者间接影响脂质代谢,从而导致脂质在肌纤维细胞中堆积而引起的一组代谢性疾病[3]。本病轻症者发病较晚,主要表现为骨骼肌无力,症状呈波动性。病情严重者发病较早,不仅累及骨骼肌,还可累及神经系统、心脏、肝脏、肾脏及消化系统等。本研究18例患者的病例特征与文献报道基本相符:所有患者均有发作性或波动性肢体无力,突出特征为不耐疲劳,远路程易疲劳,休息后缓解;38.9%累及颈肌和咀嚼、吞咽肌,表现为颈部力弱、抬头困难,声音嘶哑、饮食呛咳、咀嚼、吞咽困难;23.08%有消化道症状,如恶心、烧心、反酸,可能与胃肠道粘膜脂质沉积有关[4]。部分患者可累及心脏,表现为活动后心慌、胸闷、气短;还有1例患者伴有腰部酸软,双下肢水肿等肾脏损害,考虑为本病累及到肾脏。33%~55.6%患者受累肌肉有压痛、萎缩、腱反射减弱,可能与周围神经损害有关。由于本病临床表现复杂多样,常被误诊为多发性肌炎、重症肌无力、进行性肌营养不良、吉兰-巴雷综合征、糖原累积性肌病等。本组病例临床曾误诊为多发性肌炎者10例,进行性肌营养不良者4例,重症肌无力者1例,因此肌活检病理检查对本病的诊断至关重要。
本组病例有66.7%患者的CK高于正常,以轻度升高者占多数,也有极少数患者CK到达6000 U/L;55.6%的患者累及到肝脏,以ALT、AST增高为主。所有患者的肌电图均异常,大多数为肌源性损害,有少数为肌源性和神经源性并存。
肌肉活检病理检查有助于确诊本病。活检肌肉标本镜下观察均可见SSB染色为阳性。Gomori染色未见破碎红毛样纤维(RRF)。PAS染色未见糖原累积。NADH-TR染色可见颗粒深染,I型纤维为主。ATP染色为Ⅰ、Ⅱ型纤维呈镶嵌排列,部分区域有成簇现象,表明神经纤维有损伤。电镜下偶见坏死纤维,在肌原纤维间、肌膜下、核旁有脂肪组织浸润,未见到明显炎细胞浸润、间质水肿、结缔组织增生,小血管未见到增生,血管壁薄厚均匀一致。
对于本病的治疗,由于大部分脂质沉积性肌病是由于肉碱缺乏所致,故可采用肉碱替代治疗。有报道[5],肉碱疗法显示出明显的临床效果,是一种可开发性的治疗方案。另外,可给予小剂量肾上腺糖皮质激素治疗,以促进脂肪的利用,从而获得较好的疗效。服用 ATP、辅酶 A、辅酶Q10、B族维生素和左卡尼汀治疗也可以使多数患者的临床症状好转。另外,可低长链脂肪酸、高碳水化合物饮食,多食富含肉毒碱的牛羊肉、鱼类,可少食多餐,减少糖原转化为脂肪,避免饥饿、劳累、受寒等。
[参考文献]
[1]张艺凡,李 媛,夏 聪,等. 误诊为重症肌无力的疾病临床分析[J]. 实用医学杂志,2017,33(5):742-746.
[2]马明明,王雪晶,丁雪冰,等. 以反复跌倒发作为表现的脂质沉积性肌病临床及病理特征分析[J]. 中国实用神经疾病杂志,2014,17(23):19-21.
[3]张 宁,蔡 艳,肖 波. 脂质沉积性肌病临床和病理特征分析[J]. 中风与神经疾病杂志,2009,26(4):422-425.
[4]王 韵,刘 潇,王朝霞,等. 核黄素反应性脂质沉积性肌病与多发性肌炎临床特点对比分析[J]. 中华医学杂志,2014,94(7):503-506.
[5]Subasree R,Gayathri N,Rita C. Lipid storage myopathy with clinical markers of Marfan syndrome:A rare association[J]. Ann Indian Acad Neurol,2012,15(4):332-335.
