珠穆朗玛峰不同海拔梯度上土壤细菌和真菌群落变化特征
2018-05-16张丹丹张丽梅沈菊培
张丹丹,张丽梅,沈菊培,旺 姆
1 中国科学院生态环境研究中心 城市与区域国家重点实验室,北京 100085 2 中国科学院大学,北京 100049 3 西藏农牧学院,林芝 860000
土壤微生物数量和种类繁多,每克土壤中微生物的数量可达几十亿,上万个物种[1]。微生物是土壤最具有生命力的组分,驱动着土壤中几乎所有已知的生物反应过程,在有机物质分解、养分循环、污染物转化与分解、温室气体产生和消减等过程中起着重要作用,对土壤肥力形成、土壤生态系统的结构和功能维持乃至全球变化都具有重要影响[1- 3]。鉴于土壤微生物的重要功能及其对外界环境变化的敏感响应,土壤微生物常被用作评价环境变化的重要指标[3- 4]。
全球气候变化早已成为不争的事实,据IPCC(2007)报告预测到21世纪末,全球地表温度可能升高1.1—6.4℃[5]。高寒生态系统及干旱半干旱生态系统作为全球变化的敏感区域,主要分布在北极,北部森林,青藏高原和高山苔原等地,其气候变暖速度高于地球平均水平;此外,气候变化通过改变降水格局而加速高寒生态系统退化,影响土壤微生物的群落组成和生物多样性[6]。因此,高寒生态系统对气候变化的响应与反馈不仅影响着当地生态系统,也将对全球生态系统及人类生活产生巨大影响。近年来,人们对高寒生态系统土壤微生物展开了大量的研究。Bryant等对美国科罗拉多州落基山脉(海拔2400—3600m)细菌多样性的研究发现细菌类群丰富度、系统发育多样性均随海拔高度上升而降低[7];Margesin等发现高山(海拔2300—2530m)和亚高山(海拔1500—1900m)土壤微生物活性随海拔增加而降低,且微生物群落组成随海拔增加变化,一些嗜冷细菌、真菌和革兰氏阴性细菌主要在高海拔地区出现[8]。Shen等对我国长白山地区的研究也发现土壤细菌群落随海拔明显变化,细菌群落组成及优势门的相对丰度与土壤pH显著相关[9]。可见海拔相关因素在很大程度上影响着微生物的丰度和群落结构。
青藏高原是世界上海拔最高的高原,地势险峻多变,地形复杂,素有“世界屋脊”和“地球第三极”之称,是北半球气候变化的启张器和调节器,不仅直接驱动中国东部和西南部气候的变化,而且对北半球具有巨大的影响,甚至对全球的气候变化,也具有明显的敏感性、超前性和调节性[10]。青藏高原板块构造隆升形成了其独特的自然地理环境:高寒,辐射强,日照时间长,缺氧,养分浓度低,气温随高度和纬度的升高而降低。因此,表层土壤微生物群落在对抗上述极端环境过程中可能会形成特有的生存适应能力和群落组成差异。此外,由于青藏高原对全球气候的敏感性,全球变暖和CO2浓度升高可能会对地下微生物产生复杂的影响[11]。如芦晓飞利用克隆测序技术对西藏米拉山高寒草甸土壤(海拔3600m)细菌和古菌群落多样性发现该区域存在着丰富的特殊微生物资源,但是由于受到气候变化和人类活动影响,退化草甸土壤的微生物群落结构发生改变[12]。彭岳林等对藏北高原不同海拔(海拔4584—4956m)草地植物丛枝菌根(AM)真菌种群多样性的调查研究发现Glomus属真菌为最优势属,且草地类型、海拔高度和土壤pH值是影响AM真菌生态分布的重要因子[13]。李超男等对贡嘎山海拔梯度上(海拔1800—4100m)土壤甲烷氧化菌的研究发现土壤理化性质和气候变化综合作用是造成甲烷氧化菌群落结构和多样性变化的主要因素[14]。
以上研究虽然都是针对青藏高原地区微生物群落开展的,但是主要集中在低海拔地区,尤其在珠峰永久雪线以上的研究非常少。鉴于青藏高原在全球变化中的特殊地位和作用,深入研究不同海拔梯度下细菌和真菌物种多样性及群落组成沿海拔高度的变化及其规律,对于理解全球气候变化对青藏高原生态系统的影响和及其响应机制具有重要意义。此外,高寒生态系统因其独特的自然气候条件,赋予了一些土壤微生物的独特优势,也是微生物资源的潜在宝库,研究青藏高原珠穆朗玛峰高原土壤微生物丰度和群落组成为合理开发利用高寒生态系统微生物资源提供参考。以往研究中,发现珠穆朗玛峰高山土壤中(海拔4000—6550m)氨氧化细菌和古菌的丰度和群落组成随海拔梯度发生明显变化,尤其在雪线附近发生显著转变[15],但对于土壤中细菌和真菌群落整体组成情况不清楚。本研究利用活菌计数法(CFU)、实时定量PCR(real-time PCR)技术,并结合磷脂脂肪酸PLFA分析法,变性梯度凝胶电泳技术(DGGE)和克隆测序方法研究了青藏高原珠穆朗玛峰坡底和永久雪线以上高海拔(4000—6550m)地区土壤细菌和真菌群落的组成特征,以期为理解全球变化对青藏高原生态系统的影响以及发掘高寒地区土壤微生物资源提供基础信息。
1 材料和方法
1.1 土壤样品采集
本研究土壤样品为中国科学院第四次珠穆朗玛峰远征期间(2005年4月至6月)采集自珠峰北部斜坡。除3个样品(M1—M3)取自西藏日喀则地区的农田土壤外(28°82′N,29°28′E;海拔4000m左右,年均温6.3℃),其余样品(M4—M12)均采自珠峰北坡不同海拔高度(28°01′570″—28°08′385″N,86°51′533″—86°56′686″E)的裸地,海拔高度分别为5350,5400,5700,5850,6000,6150,6300,6450,6550m。每个采样点均取5个重复次样点,采样时用铲子移除大石头和积雪层后采集0—10cm深度的土壤或细砂砾,混合后于移动冰箱运回实验室,部分样品保存于4℃用于活菌计数和土壤理化性质分析,其余样品保存到-20℃冰箱用于DNA提取和分析。
1.2 磷脂脂肪酸(PLFA)分析
磷脂脂肪酸的提取方法和分析依照Bardgett等人描述的方法[16]。操作步骤如下:称取4g土壤样品于特氟龙管内,用氯仿-甲醇-磷酸缓冲液(1∶2∶0.8)提取总脂类,通过离心分离得到的磷脂脂肪酸经50℃碱性甲酯化后利用气体色谱Agilent 6820测定土壤微生物磷脂脂肪酸的含量,以标准定性C11到C20混合细菌酸甲酯为外源标准(Bacterial Acid Methyl Esters CP Mix, Matreya Inc.)。每个样品进行3次重复实验,脂肪酸的命名方法如Frostegard等人[17]描述。细菌生物量通过3-OH 12:0,14:0, 2-OH 14:0,i- 15:0,和a- 15:0,15:0,i- 16:0,16:1ω9c,16:0,i- 17:0,17:0cy和10Me18:0的脂肪酸进行估算[18- 21],真菌生物量用18:2ω9,12,18:1ω9c,18:1ω9t进行估算[17- 20]。此外,革兰氏阳性细菌通过i- 15:0、a- 15:0、i- 16:0,i- 17:0和10Me18:0估算,革兰氏阴性细菌利用3-OH 12:0,14:0, 2-OH 14:0,15:0,16:1ω9c,16:0和17:0cy计算[22- 24]。
1.3 活菌计数
样品活菌计数采用稀释平板法进行,为尽可能模仿原生境寡营养条件培养,细菌计数以10倍稀释的PGY培养基(PGY:蛋白胨10g,酵母膏5g,葡萄糖1g,蒸馏水1L,pH=7.4),真菌计数以含50mg/L硫酸链霉素的10倍稀释的马铃薯葡萄糖琼脂(PDA:马铃薯200g,葡萄糖20g,琼脂20g,蒸馏水1L,pH自然)培养基进行。操作过程为:称取土壤或砂砾样品10g,加入90mL无菌水后振荡30min,取1mL土壤悬浮液转移到装有9mL无菌水试管中,充分混合,并进行系列稀释。吸取不同稀释程度的菌悬液各0.1mL至培养皿中,涂布均匀。在正式进行计数培养前,选择了少量样品进行温度影响试验,发现在4℃培养一个月后细菌和真菌的(Colony forming units,CFUs)数比22℃培养明显少,故在进行正式计数培养时将平板置于22℃黑暗培养7d和14d后进行菌落计数,计算可培养微生物的数量。
1.4 DNA提取和PCR变性梯度凝胶电泳(DGGE)
土壤总DNA提取采用FastDNA SPIN Kit for Soil 试剂盒(Q·BIOgene, Inc., Carlsbad, CA)进行,所有操作依照产品说明书进行,鲜土称重0.8—1.0g,破碎速度设定为5.5m/s,破碎时间20s。提取的DNA样品在1%的琼脂糖凝胶中进行电泳检测后使用微量分光光度计Nanodrop® ND- 1000测定浓度和纯度(NanoDrop Technologies, Wilmington, DE)。
以引物f954-gc和r1369[25]和引物NS1和Fung GC[26](表1)分别对细菌16S rRNA基因和真菌18S rRNA基因进行普通PCR扩增。PCR扩增在PTC- 200 thermal cycler (MJ-Research, Waltham, MA) 进行,扩增程序如表1。PCR反应混合物的体积为50μL,包括1 × PCR buffer, 2mmol/L MgCl2, 400nM正反向引物浓度, 250μM dNTP, 2.5U ExTaqDNA polymerase (TaKaRa Bio Inc. Otsu, Shiga, Japan),0.4mg/mL牛血清白蛋白(BSA)和DNA模板10ng。所获得的PCR产物在6%聚丙烯酰胺胶中进行DGGE电泳,细菌所用的变性胶浓度范围是40%—60%,真菌的变性胶浓度范围为10%—30%。电泳在DCode Universal Detection System Instrument (Bio-Rad Laboratories, Hercules, CA, USA)进行。电泳条件为120v,60℃恒温条件,7h。依照操作说明将凝胶在1:10000 SYBR Gold 染料 (Invitrogen Molecular Probes, Eugene, USA)染色30min后利用凝胶呈像仪(G:BOX HR,Syngene, Frederick, USA)进行呈像扫描,并用Quantity One (Bio-Rad Laboratories, Hercules, USA)软件分析结果。
对DGGE图谱中清晰条带进行切胶回收,并将胶条转移到50μL 10mmol/L Tris-HCl (pH 8.0)缓冲液中,反复冻融3次以上。取上清液2μL用无GC夹子原引物再次扩增。PCR产物以胶回收试剂盒(TaKaRa Bio Inc., Shiga, Japan)切胶纯化后,连接到pGEM-T Easy Vector上(Promega, Madison, USA)试剂盒连接载体,转化大肠杆菌 JM109感受态细胞(JM109)(TaKaRa, Japan),涂布到含有氨苄青霉素(Ampicillin)/IPTG/X-Gal的LB (Luria-Bertani)培养基上,37℃下培养16—18h。随机选取若干白色克隆子,采用菌体直接扩增方式,用pGEM-T Easy Vector通用引物T7/SP6扩增外源插入片段。每个条带挑选3个阳性克隆子测序,利用DNAMAN 6.0.3.48 (Lynnon Biosoft, USA)进行序列分析,并把所得正确长度的序列提交NCBI数据库进行Blast序列对比,下载最相似菌种序列作为系统发育树的参考序列。然后采用Clustal W和Mega 4.0软件建立Neighbor-Joining系统发育树,系统发育树各分支置信度由自举分析方法(Bootstrap)检验,重复1000次。
1.5 荧光定量PCR分析(real-time PCR)
利用实时荧光定量PCR对土壤中的细菌和真菌丰度进行定量分析,所用引物和探针具体信息详见表1。细菌荧光定量PCR分析采用探针法:使用TaqMan探针TM1389(5′-CTTGTACACACCGCCCGTC- 3′)(Takara Bio Inc. Otsu, Shiga, Japan),定量引物分别为BACT1369F(5′-CGGTGAATACGTTCYCGG- 3′)/PROK1541R(5′-AAGGAGGTGATCCRGCCGCA- 3′)[27];真菌扩增使用引物ITS1f(5′-TCCGTAGGTGAACCTGCGG- 3′)/5.8s(5′-CGCTGCGTTCTTCATCG- 3′)[28]。实时荧光定量PCR反应体系为25μL,细菌16S rRNA基因的扩增使用Premix Ex TaqTM(TaKaRa),真菌ITS区扩增采用SYBR® Premix Ex TaqTM试剂盒(TaKaRa),反应条件和引物浓度参见表1。已提取的DNA经5或10倍稀释后作为模版DNA添加到混合体系中,其终浓度为1—10ng,同时加入0.4mg/mL的牛血清白蛋白(BSA)用于减少杂质对PCR反应的抑制作用。每个样品做3个平行。
1.6 数据处理
利用quantity one软件对DGGE胶图像中条带的位置和亮度进行处理,依据数字化结果,计算多样性指数。采用canonical correspondence analyses (CCAs)分析环境和土壤变量对微生物种间群落结构的影响,在CANOCO 4.5软件中进行。CFU数据和基因拷贝数经对数转换后进行ANOVA分析。本研究中的统计分析在SPSS 13.0软件中进行,其中多组数据间的方差分析采用单因素方差分析(Duncan,P<0.05);相关分析采用Spearman相关分析。
细菌和真菌rRNA基因序列GenBank号为:(递交中)
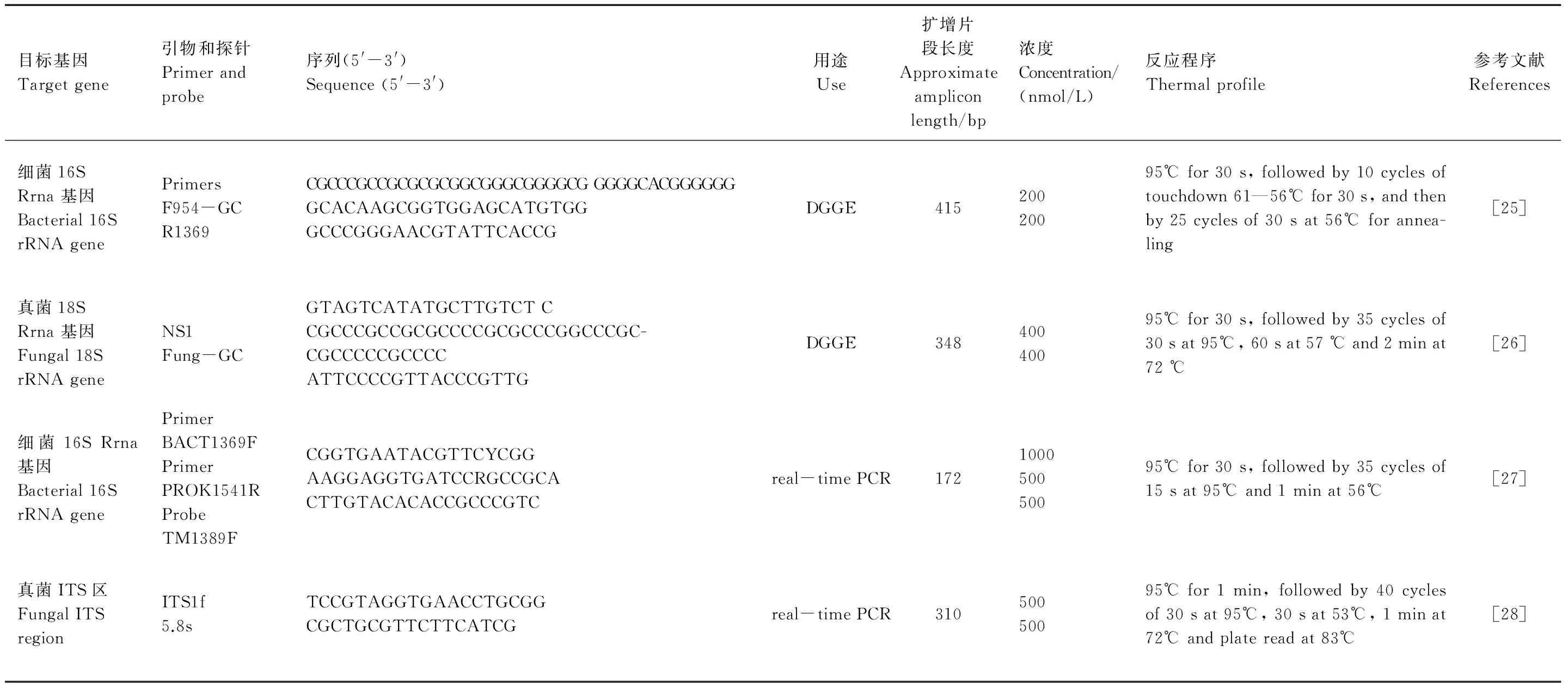
表1 本研究中用到的引物,探针和PCR条件
2 结果
2.1 土壤的基本理化性质
土壤的基本理化性质如我们之前的研究报道[15]。海拔高度在4000m左右的农田土壤(M1—M3)pH值为8.6—8.7,海拔高于5000m高度的样品(M4—M12)pH值在9.0至9.1之间(图1A)。
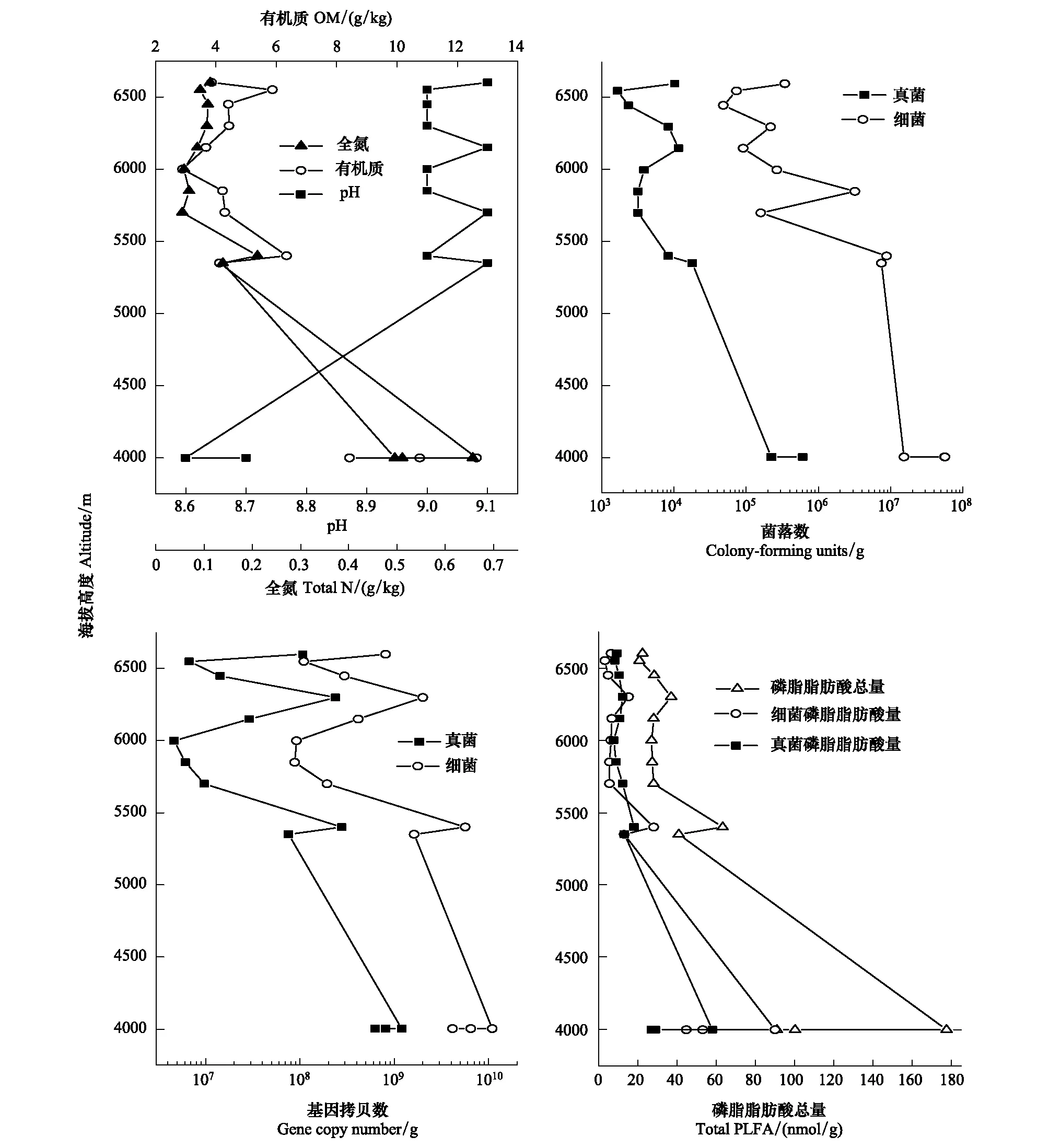
图1 青藏高原珠穆朗玛峰不同海拔梯度土壤样品的理化性质及其细菌和真菌的丰度变化Fig.1 Physicochemical properties and abundance changes of bacteria and fungi of soil samples along different altitudes from Mount Everest, on the Tibetan Plateau
在低海拔土壤样品中(M1—M3,4000m),有机质含量范围在8.4—12.6g/kg干土之间,土壤全氮在0.51—0.66g/kg干土之间。而高海拔土壤样品(M4—M12,≥5350m)土壤有机质和全氮明显低于低海拔土壤样品,其有机质含量为2.9—6.3g/kg干土,全氮为0.06—0.21g/kg干土(图1)。
2.2 细菌/真菌活菌计数和16S rRNA基因丰度
利用10倍稀释的PGY和PDA培养基对细菌和真菌进行的平板计数分析结果如图1所示。样品中可培养细菌数量随海拔增加而降低,在低海拔农田土壤样品(M1—M3)中达每克干土含(1.53—5.68)×107CFU/g,高海拔土壤/砂砾样品(M4—M12,≥5350m)(每克干重含7.36×104—8.83×106CFU/g)高1到2个数量级。所有土壤中可培养真菌的数量均较细菌低2个数量级,但变化趋势与细菌相似,在低海拔农田土壤中为每克干土(2.21—6.09)×105CFU/g之间,高海拔样品(M4—M12,≥5350m)中为1.67×103—1.16×104CFU/g干土。相关分析结果表明,细菌和真菌的CFU均与海拔高度呈负相关(r1=-0.797,r2=-0.656,P<0.05),与全氮含量呈显著正相关(r1=0.534,r2=0.565,P<0.05)。
利用实时荧光定量PCR技术对细菌的16S rRNA基因丰度和真菌ITS丰度进行的分析结果表明,细菌16S rRNA基因丰度随海拔高度增加而降低,在低海拔农田土壤样品(M1—M3)达4.13×109—1.09×1010拷贝数/g干土,高于高海拔样品(M4—M12,≥5350m)(8.82×107—5.63×109拷贝数/g干土)1到2个数量级;所有土壤真菌ITS丰度较细菌低1个数量级,但其变化趋势与细菌相类似,在低海拔土壤样品中丰度范围在6.27×108拷贝数/g干土至1.20×109拷贝数/g干土之间,在高海拔土壤样品中丰度在4.63×106—2.77×108拷贝数/g干土之间变化(图1)。与活菌计数结果类似,细菌的rRNA基因和真菌ITS的丰度与土壤海拔高度呈显著负相关(r1=-0.481,r2=-0.543,P<0.05),与土壤有机质(r1=0.606,r2=0.636,P<0.05)和全氮(r1=0.667,r2=0.697,P<0.05)呈正相关(表2)。
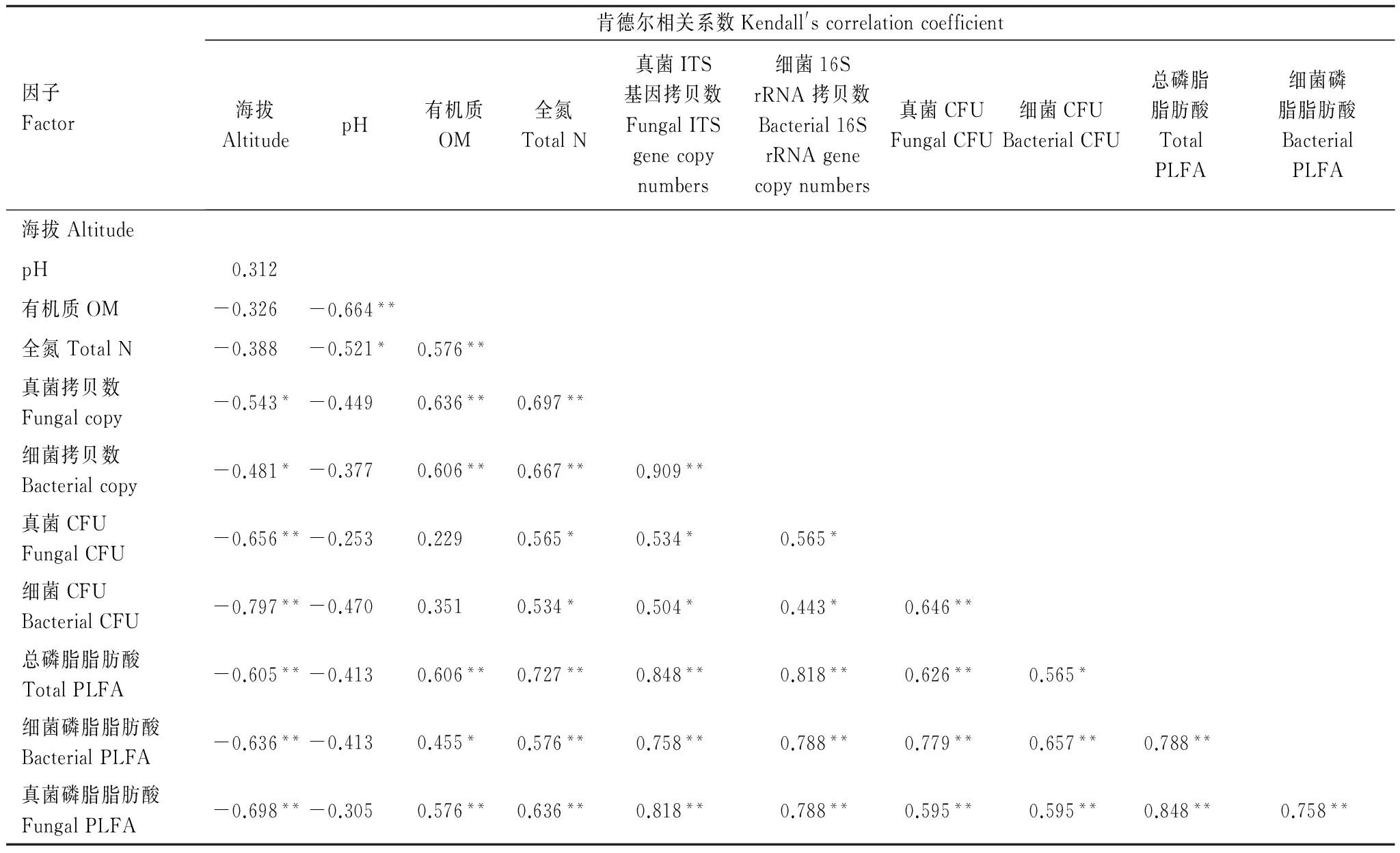
表2 土壤基本属性与多种微生物多样性相关分析
2.3 细菌和真菌磷脂脂肪酸含量和组分
磷脂脂肪酸分析结果表明,不同海拔梯度样品的磷脂脂肪酸总量(total PLFA)在20.86和177.58nmol/g之间,其中细菌来源的磷脂脂肪酸含量在8.87—108.42nmol/g之间,真菌来源的为7.82—58.16nmol/g,无论是总的PLFA含量,还是细菌和真菌的PLFA含量均随着海拔增加而降低(图1,图2),与海拔呈显著负相关(P<0.05),与有机质和全氮含量呈显著正相关(P<0.05)(表2)。
在供试的12个土壤样品中,共检测到15种C12—C18的PLFAs组分,所有样品中均检测到饱和脂肪酸(3-OH 12:0,14:0, 2-OH 14:0,i- 15:0, a- 15:0,15:0,i- 16:0, 16:0,i- 17:0和10Me18:0)、单不饱和脂肪酸、多不饱和脂肪酸和环丙烷脂肪酸。随着海拔高度增加,样品中PLFA种类和数量逐渐降低,其中低海拔土壤样品(M1—M3)含有15个单体PLFAs,M4和M5其次,含有12个单体PLFAs,而在海拔高度高于5700m的样品中(M6—M12),仅含有7种PLFAs,但这7种PLFAs(2-OH 14:0, 16:1ω9c, 16:0, 18:0,18:2ω9,12, 18:1ω9c, 18:1ω9t)在所有海拔高度样品中均有分布,其中2-OH 14:0, 16:1ω9c, 16:0代表革兰氏阴性细菌的特征脂肪酸[22-23],18:2ω9,12, 18:1ω9c和18:1ω9t代表真菌的特征脂肪酸[22-23]。而代表革兰氏阳性细菌的i- 15:0,i- 17:0和代表革兰氏阴性细菌的17:0△在海拔大于5350m的样品(M4—M5)中未检测到,代表革兰氏阴性细菌的3-OH 12:0,14:0,15:0和代表革兰氏阳性细菌的i- 15:0和i- 16:0的组份在海拔≥5700m的样品(M6—M12)中均未检测到(图2),表明这些微生物类群不能适应于高海拔环境。
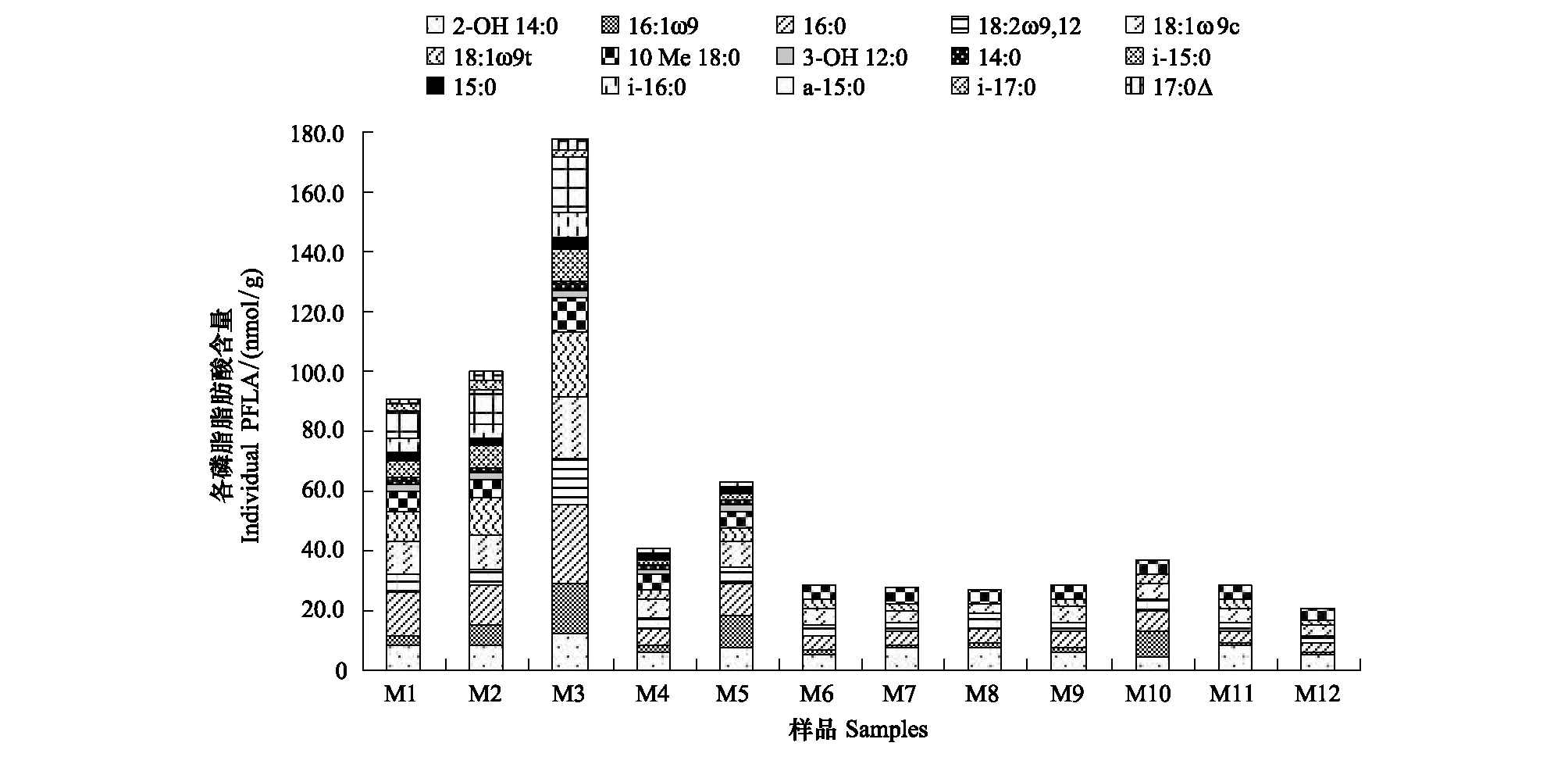
图2 青藏高原珠穆朗玛峰不同海拔梯度样品中的PLFA种类分布Fig.2 The content of PLFA composition in samples from Mount Everest, on the Tibetan PlateauM1—M3代表低海拔农田土壤样品(4000m),M4—M12代表高海拔土壤样品(≥5350m)
对磷脂脂肪酸PLFAs组成与环境变量进行的典范对应分析(Canonical correspondence analysis)结果显示,具有相似海拔高度的样品聚集在一起,其中,低海拔农田土壤样品(M1—M3)和高海拔土壤样品(M4—M12)沿y轴分开,而来自最高海拔的样品(M10—M12)与海拔稍低的样品(M4—M9)沿x轴被分开,x轴和y轴分别解释了87.2%和7.4%的变异。此外,海拔高度、土壤有机质和全氮对PLFAs的组成影响显著(P<0.05)(图3)。
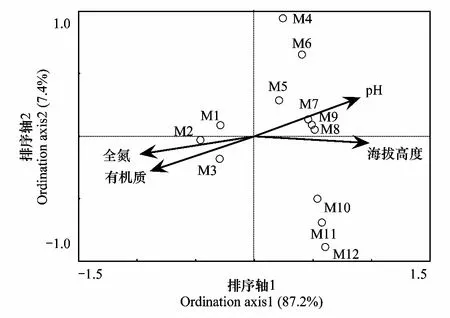
图3 微生物磷脂脂肪酸群落组成与环境变量的典范对应分析(CCA) Fig.3 Canonical correspondence analysis of PLFA composition and environmental variablesM1—M3代表低海拔农田土壤样品(4000m),M4—M12代表高海拔土壤样品(≥5350m)
2.4 不同海拔梯度样品细菌和真菌群落组成
对细菌16S rRNA基因和真菌18S rRNA基因的变性梯度凝胶电泳(DGGE)后,利用quantity one 软件对DGGE胶图像中条带的位置和亮度进行提取后,进行UPGMA聚类分析结果显示,细菌UPGMA聚类分布图中低海拔农田土样(M1—M3)聚成一个小支,与高海拔样品明显分开(图4),表明低海拔农田土壤和高海拔样品中细菌群落组成明显不同。对于真菌来说,则只观察到高海拔样品(M10—M12)单独聚集(图5)。总的来说,低海拔农田土壤样品(M1—M3)DGGE图谱更相似,而在高海拔土壤样品(M4—M12,≥5350m)中,细菌和真菌条带数量均在M5样品中最高。
选择细菌DGGE图谱中代表低海拔土壤样品M1和代表高海拔土壤样品M4、M5、M6和M9清晰条带进行克隆测序并构建细菌系统发育树(图6),结果发现,在M1样品中检测到变形菌纲(Proteobacteria)、酸杆菌纲(Acidobacteria)和放线菌纲(Actinobacteria),M4样品中检测到变形菌纲(Proteobacteria),M5中检测到变形菌纲(Proteobacteria),芽单胞菌纲(Gemmatimonadetes),蓝藻菌纲(Cyanobacteria),异常球菌属(Deinococcus-Thermus),厚壁菌门(Fimicutes),装甲菌门(Armatimonadetes),拟杆菌纲(Bacteroidetes)和放线菌纲(Actinobacteria),M6中检测到放线菌纲(Actinobacteria),M9中检测到酸杆菌纲(Acidobacteria),其中5个样品中处于同一位置的DGGE条带测序结果一致。从图6可以看到,所有土样中均含有区域1(条带24)和区域3(条带10)的条带,测序结果显示这两个区域的条带代表变形菌纲(Proteobacteria),说明变形菌是青藏高原地区的优势菌群;区域2条带6和25代表芽单胞菌纲(Gemmatimonadetes)仅分布在高海拔样品中;此外,区域4条带3代表的放线菌纲(Actinobacteria)主要分布在低海拔样品中,在高海拔样品中较为少见。由于真菌DGGE图谱中不同海拔高度土壤样品清晰条带较少,将所有清晰条带克隆测序并构建了真菌系统发育树(图7),结果显示真菌序列分布在子囊菌门(Ascomycetes)、担子菌门(Basidiomycota)和其他真核生物(Cercozoa)三个簇中;结合DGGE图谱可以看出区域1条带4,5和30代表Cercozoa仅分布在高海拔样品中,而区域2条带17,20代表子囊菌门(Ascomycetes)在所有土样中均有分布,说明子囊菌门(Ascomycetes)为青藏高原地区的优势菌群(图7)。

图4 青藏高原珠穆朗玛峰不同海拔梯度下土壤样品中细菌DGGE图谱及其UPGMA聚类分布Fig.4 Denaturing gradient gel electrophoresis (DGGE) profiles and the UPGMA dendrogram of bacteria in soils from Mount Everest, on the Tibetan Plateau

图5 青藏高原珠穆朗玛峰不同海拔梯度下土壤样品中真菌DGGE图谱及其UPGMA聚类分布Fig.5 Denaturing gradient gel electrophoresis (DGGE) profiles and the UPGMA dendrogram of fungi in soils from Mount Everest, on the Tibetan Plateau
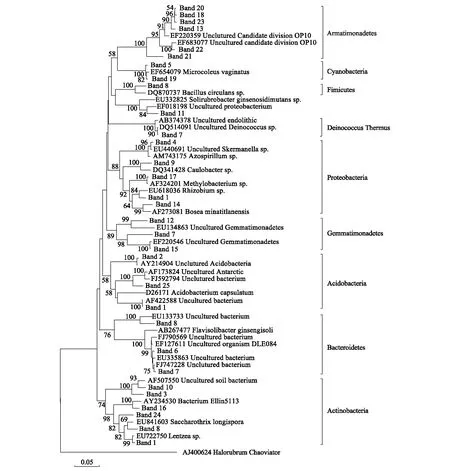
图6 细菌16S rRNA基因系统发育树Fig.6 Phylogenetic tree of bacterial 16S rRNA gene in soils from Mount Everest, on the Tibetan Plateau
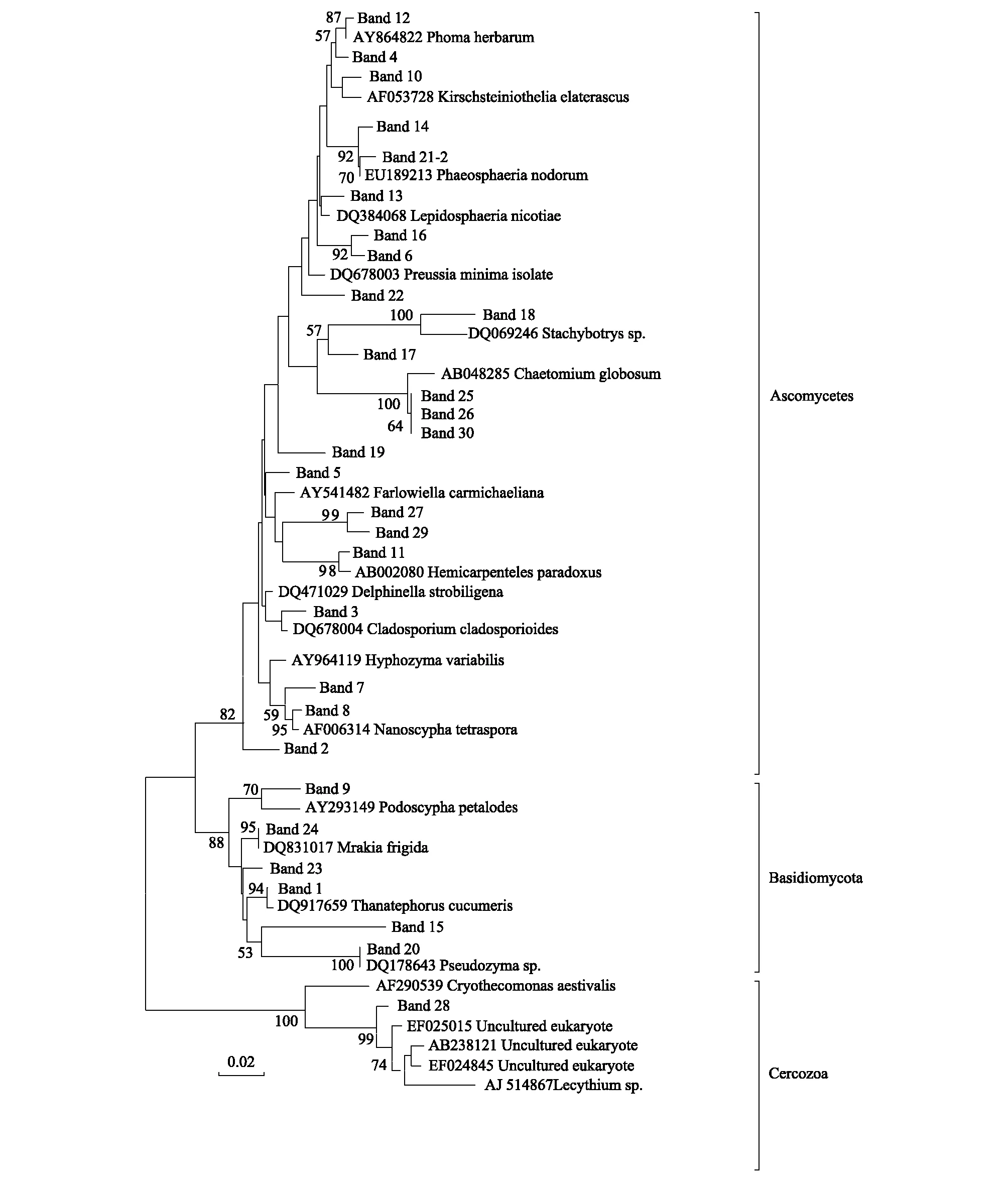
图7 真菌ITS基因系统发育树Fig.7 Phylogenetic relationships among fungal sequences in soils from Mount Everest, on the Tibetan Plateau
3 讨论
3.1 细菌和真菌数量随海拔增加的变化特征
本研究中,用10倍稀释的细菌和真菌培养基进行的活菌计数结果表明低海拔土壤样品细菌CFU数(1.53—5.68)×107CFU/g明显高于高海拔样品(7.36×104—8.83×106CFU/g)。类似地,真菌CFU分布情况与细菌一致,低海拔土壤样品CFU(2.21—6.09×105CFU/g)较高海拔样品高1—2个数量级(1.67×103—1.16×104CFU/g)。利用荧光定量PCR技术对细菌16S rRNA和真菌ITS基因丰度的分析结果与活菌和真菌计数结果一致,低海拔土壤样品细菌和真菌丰度明显高于高海拔地区土壤样品1到2个数量级。相关分析结果表明无论细菌和真菌的CFU数量,还是16S rRNA和ITS基因拷贝数均与全氮和有机质显著正相关,与海拔高度显著负相关性。此外,细菌和真菌PLFA量随海拔增加而降低,与土壤海拔呈负相关,与有机质和全氮呈显著正相关。随海拔高度增加,土壤养分、氧气、温度降低,辐射增加,这些都可能是导致微生物生长代谢减弱、生物量随海拔增加而减少的决定性因素。结果与以往在青藏高原腹地海拔高度在3400—4300m的三江源自然保护区高寒草原的研究结果一致,说明即使在严酷的生存环境下,青藏高原仍存有丰富的微生物类群,且高海拔高寒等极端环境明显抑制了微生物的活动从而导致其微生物量逐渐降低[31- 32]。此外,CFU和荧光定量PCR结果显示青藏高原珠穆朗玛峰土壤样品中细菌丰度明显高于真菌丰度一个数量级,且细菌和真菌变化趋势呈正相关关系(r=0.909,P<0.05),这与任佐华等人对青藏高原高寒草地土壤(海拔3400—4200m)微生物活性和微生物量的研究结果相类似[31-33],导致这种现象的原因可能是细菌和真菌占据的生态位相似,在青藏高原极端生存环境下,细菌由于个体小,繁殖快在资源竞争中处于优势[34- 36]。
3.2 不同海拔梯度上细菌和真菌的群落组成特征
磷脂脂肪酸(PLFA)常用来表征复杂环境中具有活性的微生物群落组成。在12个供试样品中共检测到15种PLFAs,包括代表革兰氏阳性细菌的脂肪酸组份i- 15:0、a- 15:0、i- 16:0和i- 17:0,代表革兰氏阴性细菌的组份3-OH 12:0,14:0, 2-OH 14:0,15:0,16:1ω9c,16:0,17:0△和10Me 18:0,和代表真菌的组份18:2ω9,12,18:1ω9c,18:1ω9t。无论是细菌还是真菌的脂肪酸组分均随海拔增加而减少,主要表现为海拔高度大于5350m(雪线)时,表征革兰氏阳性细菌的脂肪酸(a- 15:0和i- 17:0)未被检测到,当海拔高度增加到5700m以上时,表征革兰氏阳性细菌的所有脂肪酸(i- 15:0、a- 15:0、i- 16:0和i- 17:0)均未被检测到。同时,表征革兰氏阴性细菌的脂肪酸17:0△在海拔高度大于5350m时未被检测到,当海拔增加到5400m时,3-OH12:0和14:0也未被检测到,但2-OH 14:0,16:1ω9c和16:0脂肪酸在全部样品中均有分布,说明青藏高原珠穆朗玛峰土壤中细菌多样性随海拔增加而降低,且革兰氏阳性细菌对海拔高度的变化较革兰氏阴性细菌更为敏感,可能的原因是革兰氏阳性细菌细胞壁结构简单,只含有90%肽聚糖和10%磷壁酸,其对抗外界环境变化的缓冲能力较弱,而革兰氏阴性细菌细胞壁较为复杂,不仅含有肽聚糖,其细胞壁外还有由脂多糖、磷脂和脂蛋白等若干种蛋白质组成的外膜能够有效的阻止或减缓有害物质或恶劣环境对细胞的迫害,从而保护细胞的正常生长代谢[37]。此外,表征真菌的PLFAs(18:2ω9,12,18:1ω9c,18:1ω9t)在所有土壤样品中均有分布,说明海拔高度增加未能影响真菌在高山土壤的分布,与蔡晓布等在西藏高原草地生态系统中观察到的结果一致[38],这可能是由于真菌在生理上更耐受低温,以及其通过产生孢子抵御不良环境等所致。该结果与DGGE图谱的UPGMA聚类分析结果相一致,再次表明不同海拔高度能够显著影响细菌群落组成,但对真菌群落变化没有显著影响。
对微生物磷脂脂肪酸PLFA组成与环境变量的典范对应分析(Canonical correspondence analysis)结果表明海拔高度、土壤有机质和全氮是决定极端环境下高山土壤微生物群落的组成和多样性的主要因子。与对微生物量的影响相似,随着海拔上升,植被逐渐退化,土壤养分减少,不仅影响土壤微生物数量,也会对土壤中微生物类群造成极大的危害,导致土壤微生物多样性降低[32]。与此同时,高山温度会随海拔升高而降低,Lipson等人针对不同季节和海拔梯度下细菌群落结构与温度的关系研究中发现,低海拔地区微生物群落对温度较高海拔地区更为敏感[8,39]。
对细菌DGGE主要条带的克隆测序分析结果显示,变形菌纲(Proteobacteria)在低海拔和高海拔样品中均有分布,说明变形菌是青藏高原土壤的优势菌群;芽单胞菌仅出现在了高海拔样品中,这可能与芽单胞菌易受环境湿度的影响有关[40]。DeBruyn等通过对南极旱地土壤、高山干旱土壤和高山苔原等13个不同类型土壤中芽单胞菌丰度和群落组成的研究发现,芽单胞菌相对丰度与水分负相关,芽单胞菌适宜在较为干燥的环境中生存[41]。本研究中低海拔样品主要分布在日喀则地区,其降水含量较高海拔样品丰沛,相对较高的降水量会使低海拔样品的芽单胞菌代谢受到抑制,而高海拔相对干燥的环境促进了芽单胞菌的生长繁殖,使芽单胞菌成为高海拔样品中的优势菌群。放线菌纲(Actinobacteria)主要分布在低海拔样品中出现,较少在高海拔样品中发现,说明放线菌随着海拔高度的增加而减少,这与以往对西藏土壤放线菌研究结果相似,即放线菌数量随海拔升高,冻结期增长显著减少[42]。
对真菌DGGE主要条带的克隆测序结果显示子囊菌门(Ascomycetes)广泛分布在低海拔和高海拔样品中,在4000m海拔样品中也检测到部分担子菌门(Basidiomycota),但子囊菌门(Ascomycetes)是主要优势菌群。此外,利用真菌引物NS1/Fung GC还回收到部分与丝足虫类(Cercozoa)原生生物如Cryothecomonas,Lecythium等高度相似的序列,但这些序列仅分布在高海拔冰雪覆盖的样品中,表明这些序列所代表的生物具有较强的耐寒能力,与以往的研究相类似。如Cryothecomonas最早在南极海冰和冰缘区被发现[43- 44],并相继在冰芯,沉积物和水柱[45- 48]中被发现。此外,Thaler等利用特异性荧光原位杂交技术对北极和亚北极海域中的Cryothecomonas研究发现Cryothecomonas丰度与冰和融雪量显著正相关,说明Cryothecomonas可以作为北极冰柱融化的敏感指标[48]。虽然本研究选择DGGE图谱测序的代表条带可能不会完全反映土壤中微生物群落结构,但至少代表了样品中优势微生物类群的变化特征。
4 结论
本文综合采用平板活菌计数,定量PCR,PLFA和DGGE技术探讨了青藏高原珠穆朗玛峰北坡从坡底牧草和农业土壤,到海拔6550m高度的高山土壤或风化不完全的砂砾样品中微生物的生物量和群落组成特征。结果表明受海拔依赖的相关因子,如养分(有机质、总氮)、温度等的影响,无论是活性细菌和真菌的CFU数,细菌16S rRNA和真菌ITS基因丰度,还是PLFA所代表的微生物量和物种多样性均随海拔升高明显降低。此外,PLFA中代表革兰氏阳性细菌的组分在高于5350m以上的样品中缺失,而代表革兰氏阴性细菌的部分组分和真菌组分在所有样品中都有分布,表明革兰氏阳性细菌比革兰氏阴性细菌和真菌对海拔梯度变化更为敏感,该结果与不同样品中细菌DGGE指纹图谱随海拔高度聚集成簇,而真菌DGGE指纹图谱随海拔高度变化无明显聚集的结果相一致,表明海拔高度显著影响细菌群落组成,但对真菌群落变化没有显著影响。此外,对细菌和真菌DGGE主要条带的回收测序分析结果显示变形菌纲是珠峰不同海拔高度土壤/砂砾样品中的优势菌群,芽单胞菌是高海拔样品中的优势菌群,而放线菌主要分布在低海拔样品中;真菌以子囊菌门为优势菌群。此外,利用真菌的引物还在高海拔冰雪覆盖样品回收到丝足虫类原生生物,代表了冰雪覆盖样品中所特有真核生物类群。这些结果为认识全球变化对高寒区生态系统的影响以及发掘高寒地区土壤微生物资源提供了重要信息。
参考文献(References):
[1] 傅伯杰. 中国学科发展战略: 土壤生物学. 北京: 科学出版社, 2016.
[2] 黄昌勇. 土壤学: 面向21世纪课程教材. 北京: 中国农业出版社, 2000.
[3] Nannipieri P, Ascher J, Ceccherini M T, Landi L, Pietramellara G, Renella G. Microbial diversity and soil functions. European Journal of Soil Science, 2003, 54(4): 655- 670.
[4] Romaniuk R, Giuffré L, Costantini A, Nannipieri P. Assessment of soil microbial diversity measurements as indicators of soil functioning in organic and conventional horticulture systems. Ecological Indicators, 2011, 11(5): 1345- 1353.
[5] Djukic I, Zehetner F, Mentler A, Gerzabek M H. Microbial community composition and activity in different Alpine vegetation zones. Soil Biology and Biochemistry, 2010, 42(2): 155- 161.
[6] Walker D A, Jia G J, Epstein H E, Raynolds M K, Chapin III F S, Copass C, Hinzman L D, Knudson J A, Maier H A, Michaelson G J, Nelson F, Ping C L, Romanovsky V E, Shiklomanov N. Vegetation-soil-thaw-depth relationships along a low-arctic bioclimate gradient, Alaska: synthesis of information from the ATLAS studies. Permafrost and Periglacial Processes, 2003, 14(2): 103- 123.
[7] Bryant J A, Lamanna C, Morlon H, Kerkhoff A J, Enquist B J, Green J L. Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(S1): 11505- 11511.
[8] Margesin R, Jud M, Tscherko D, Schinner F. Microbial communities and activities in alpine and subalpine soils. FEMS Microbiology Ecology, 2009, 67(2): 208- 218.
[9] Shen C C, Xiong J B, Zhang H Y, Feng Y Z, Lin X G, Li X Y, Liang W J, Chu H Y. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biology and Biochemistry, 2013, 57: 204- 211.
[10] Wu S H, Yin Y H, Zheng D, Yang Q Y. Climatic trends over the Tibetan Plateau during 1971- 2000. Journal of Geographical Sciences, 2007, 17(2): 141- 151.
[11] Xu Z H, Chen C R, He J Z, Liu J X. Trends and challenges in soil research 2009: linking global climate change to local long-term forest productivity. Journal of Soils and Sediments, 2009, 9(2): 83- 88.
[12] 芦晓飞. 西藏米拉山高寒草甸土壤微生物多样性研究[D]. 北京: 中国农业科学院, 2009.
[13] 蔡晓布, 彭岳林. 西藏高原不同海拔区域丛枝菌根真菌群落的变化. 应用生态学报, 2015, 26(9): 2803- 2810.
[14] 李超男, 李家宝, 李香真. 贡嘎山海拔梯度上不同植被类型土壤甲烷氧化菌群落结构及多样性. 应用生态学报, 2017, 28(3): 805- 814.
[15] Zhang L M, Wang M, Prosser J I, Zheng Y M, He J Z. Altitude ammonia-oxidizing bacteria and archaea in soils of Mount Everest. FEMS Microbiology Ecology, 2009, 70(2): 208- 217.
[16] Bardgett R D, Hobbs P J, Frostegård Å. Changes in soil fungal: bacterial biomass ratios following reductions in the intensity of management of an upland grassland. Biology and Fertility of Soils, 1996, 22(3): 261- 264.
[17] Frostegård Å, Bååth E, Tunlio A. Shifts in the structure of soil microbial communities in limed forests as revealed by phospholipid fatty acid analysis. Soil Biology and Biochemistry, 1993, 25(6): 723- 730.
[18] Kemp P F, Cole J J, Sherr B F, Sherr E B. Handbook of Methods in Aquatic Microbial Ecology. Florida: Lewis Publishers, 1993: 271- 284.
[19] 颜慧, 蔡祖聪, 钟文辉. 磷脂脂肪酸分析方法及其在土壤微生物多样性研究中的应用. 土壤学报, 2006, 43(5): 851- 859.
[20] Tunlid A, White D C. Biochemical analysis of biomass, community structure, nutritional status, and metabolic activity of microbial communities in soil // Stotzky G, Bollag J M, eds. Soil Biochemistry. New York, Basel, Hong Kong: Marcel Dekker Inc., 1992, 7: 229- 262.
[21] Harwood J L, Russell N J. Lipids in Plants and Microbes. Netherlands: Springer, 1984.
[22] Stahl P D, Klug M J. Characterization and differentiation of filamentous fungi based on fatty acid composition. Applied and Environmental Microbiology, 1996, 62(11): 4136- 4146.
[23] Klamer M, Bååth E. Estimation of conversion factors for fungal biomass determination in compost using ergosterol and PLFA 18: 2ω6, 9. Soil Biology and Biochemistry, 2004, 36(1): 57- 65.
[24] Schelble R T, McDonald G D, Hall J A, Nealson K H. Community structure comparison using FAME analysis of desert varnish and soil, Mojave Desert, California. Geomicrobiology Journal, 2005, 22(7/8): 353- 360.
[25] Yu Z T, Morrison M. Comparisons of different hypervariable regions ofrrsgenes for use in fingerprinting of microbial communities by PCR-Denaturing gradient gel electrophoresis. Applied and Environmental Microbiology, 2004, 70(8): 4800- 4806.
[26] May L A, Smiley B, Schmidt M G. Comparative denaturing gradient gel electrophoresis analysis of fungal communities associated with whole plant corn silage. Canadian Journal of Microbiology, 2001, 47(9): 829- 841.
[27] Suzuki M T, Taylor L T, Delong E F. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Applied and Environmental Microbiology, 2000, 66(11): 4605- 4614.
[28] Fierer N, Jackson J A, Vilgalys R, Jackson R B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Applied and Environmental Microbiology, 2005, 71(7): 4117- 4120.
[29] Hu Y J, Xiang D, Veresoglou S D, Chen F L, Chen Y L, Hao Z P, Zhang X, Chen B D. Soil organic carbon and soil structure are driving microbial abundance and community composition across the arid and semi-arid grasslands in northern China. Soil Biology and Biochemistry, 2014, 77(1): 51- 57.
[30] 吴愉萍. 基于磷脂脂肪酸(PLFA)分析技术的土壤微生物群落结构多样性的研究[D]. 杭州: 浙江大学, 2009.
[31] 任佐华, 张于光, 李迪强, 肖启明, 蔡重阳. 三江源地区高寒草原土壤微生物活性和微生物量. 生态学报, 2011, 31(11): 3232- 3238.
[32] 林超峰, 陈占全, 薛泉宏, 来航线, 陈来生, 张登山. 青海三江源区植被退化对土壤养分和微生物区系的影响. 应用与环境生物学报, 2007, 13(6): 788- 793.
[33] 曾智科. 三江源区高寒草甸土壤微生物季节动态及对草地退化的响应[D]. 西宁: 青海师范大学, 2009.
[34] Frey S D, Knorr M, Parrent J L, Simpson R T. Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. Forest Ecology and Management, 2004, 196(1): 159- 171.
[35] Wallenstein M D, McNulty S, Fernandez I J, Boggs J, Schlesinger W H. Nitrogen fertilization decreases forest soil fungal and bacterial biomass in three long-term experiments. Forest Ecology and Management, 2006, 222(1/3): 459- 468.
[36] Janssens I A, Dieleman W, Luyssaert S, Subke J A, Reichstein M, Ceulemans R, Ciais P, Dolman A J, Grace J, Matteucc G, Papale D, Piao S L, Schulze E D, Tang J, Law B E. Reduction of forest soil respiration in response to nitrogen deposition. Nature Geoscience, 2010, 3(5): 315- 322.
[37] 沈萍, 陈向东. 微生物学(第八版). 北京: 高等教育出版社, 2016.
[38] 蔡晓布, 彭岳林, 冯固, 钱成. 西藏高原草地植物AM真菌多样性及其环境影响因子研究. 土壤学报, 2005, 42(4): 642- 651.
[39] Lipson D A. Relationships between temperature responses and bacterial community structure along seasonal and altitudinal gradients. FEMS Microbiology Ecology, 2007, 59(2): 418- 427.
[40] Fawaz M N. Revealing the ecological role ofGemmatimonadetesthrough cultivation and molecular analysis of agricultural soils[D]. Knoxville: University of Tennessee, 2013.
[41] DeBruyn J M, Nixon L T, Fawaz M N, Johnson A M, Radosevich M. Global biogeography and quantitative seasonal dynamics ofGemmatimonadetesin soil. Applied and Environmental Microbiology, 2011, 77(17): 6295- 6300.
[42] 薛泉宏, 谭志远, 朱铭莪, 张晓琳. 西藏土壤放线性菌初步研究. 西北农业大学学报, 1999, 27(1): 28- 32.
[43] Garrison D L, Buck K R. The biota of Antarctic pack ice in the Weddell Sea and Antarctic Peninsula regions. Polar Biology, 1989, 10(3): 211- 219.
[44] Garrison D L, Buck K R. Protozooplankton in the Weddell Sea, Antarctica: abundance and distribution in the ice-edge zone. Polar Biology, 1989, 9(6): 341- 351.
[45] Sime-Ngando T, Juniper S K, Demers S. Ice-brine and planktonic microheterotrophs from Saroma-ko Lagoon, Hokkaido (Japan): quantitative importance and trophodynamics. Journal of Marine Systems, 1997, 11(1/2): 149- 161.
[46] Thomsen H A, Buck K R, Bolt P A, Garrison D L. Fine structure and biology ofCryothecomonasgen. nov. (Protista incertae sedis) from the ice biota. Canadian Journal of Zoology, 1991, 69(4): 1048- 1070.
[47] Tillmann U, Hesse K J, Tillmann A. Large-scale parasitic infection of diatoms in the Northfrisian Wadden Sea. Journal of Sea Research, 1999, 42(3): 255- 261.
[48] Thaler M, Lovejoy C. Distribution and diversity of a protist predatorCryothecomonas(Cercozoa) in Arctic marine waters. Journal of Eukaryotic Microbiology, 2012, 59(4): 291- 299.
