SNP位点rs6858698对慢性淋巴细胞白血病易感性的影响
2018-05-16武佳玉王晓月
武佳玉,刘 松,王晓月
(中国医学科学院基础医学研究所 北京协和医学院基础学院 生物化学与分子生物学系,北京 100005)
全基因组关联分析研究(genome-wide associa-tion study, GWAS)已经鉴定了约1 000多个与癌症发生风险相关的单核苷酸多态性(single nucleotide polymorphisms, SNP)位点,其中有很多SNP位于非编码区[1]。目前的研究表明,非编码区的SNP位点可以通过影响转录因子的结合,发挥调控活性,调节靶基因的表达[2]。然而,对于大部分非编码区SNP位点在癌症发生中的作用机制则尚不清楚,值得深入研究。
rs6858698是一个与慢性淋巴细胞白血病发生风险相关的SNP位点[3]。该SNP位于钙/钙调素依赖蛋白激酶2D(calcium/calmodulin-dependent protein kinase type Ⅱ subunit delta,CAMK2D)基因上游,距离CAMK2D的转录起始位点的长度约有760个碱基对,位于DNA酶高敏感位点内,并且具有增强子特异的组蛋白修饰信号,如组蛋白3赖氨酸27乙酰化(H3K27ac)。DNA元件百科全书(encyclopedia of DNA elements, ENCODE)数据库也显示rs6858698位于转录因子结合的热点位置[4]。鉴于rs6858698位点所在区域具有潜在的调控元件活性,本研究主要致力于探索rs6858698位点在调控基因表达方面的作用。
1 材料与方法
1.1 材料
1.1.1 细胞系:人胚肾细胞系HEK293T购自中国医学科学院基础医学研究所细胞资源中心。
1.1.2 主要试剂:DMEM培养基和胎牛血清(Hyclone公司);lentiCRISPRv2质粒、lentiGuide-Puro质粒、病毒包装辅助质粒psPAX2和PMD2.G(Addgene公司);限制性内切酶BsmBⅠ、Neon电转试剂盒、Trizol试剂、反转录试剂盒和嘌呤霉素(赛默飞世尔科技公司);T4连接酶、限制性内切酶KpnⅠ、BglⅡ和Gibson组装酶(NEB公司);琼脂糖凝胶回收试剂盒、DNA产物回收试剂盒和质粒小提试剂盒(天根生化科技有限公司);萤火虫荧光素酶质粒(pGL4.23-SCP1)、海参荧光素酶质粒(Tk)、荧光素酶报告基因检测试剂盒(Promega公司);Neofect转染试剂(零客创智生物科技公司);实时定量PCR检测试剂盒(Roche公司)。
1.2 方法
1.2.1 含rs6858698区域的报告基因载体的构建:限制性内切酶KpnⅠ和BglⅡ双酶切pGL4.23-SCP1质粒载体。同时以HEK293T基因组DNA为模板,扩增rs6858698位点附近长度约为500个碱基对的DNA序列。将PCR产物通过Gibson组装连接到pGL4.23-SCP1质粒SCP1启动子的上游,测序得到SNP位点的基因型。以测序成功的质粒为模板,进行点突变PCR,构建定点突变的质粒克隆。PCR引物见表1
1.2.2 双荧光素酶报告基因检测实验:按照1.5×105/孔将细胞接种于24孔细胞培养板中,待细胞增殖至60%~80%汇合后进行转染。每孔转染20 ng海肾荧光素酶质粒和500 ng pGL4.23-SCP1质粒(rs6858698-C和rs6858698-G),同时共转20 ng海肾荧光素酶质粒和500 ng pGL4.23-SCP1质粒空载体作为阴性对照组。24 h后检测荧光素酶活性。
1.2.3 点突变细胞株构建:选取切割位置距离rs6858698位点最近的单链导向RNA(single guide RNA, sgRNA),将其构建到lentiCRISPRv2质粒载体上。同源重组的DNA模板是一条长度为127个碱基的单链寡核苷酸[5]。使用Neon电转仪将4 μg构建成功的lentiCRISPR-v2质粒和1 μg模板DNA转染进HEK293T细胞内。48 h后加入嘌呤霉素至终浓度为1.5 μg/mL进行筛选,分选细胞单克隆,测序鉴定。
1.2.4 RNA-seq样品制备及高通量测序:将野生型细胞(rs6858698-G)和突变型细胞(rs6858698-C)分别接种于6孔细胞培养板中,每组有3个复孔。待细胞增殖至80 %汇合后,Trizol法提取细胞总RNA。由诺禾致源公司进行后续建库及全转录组测序分析。
1.2.5 实时定量PCR验证RNA-seq的分析结果:Trizol法提取细胞总RNA,取3 μg RNA为模板反转录成cDNA。实时定量PCR反应体系为1 μL cDNA,1 μL上下游引物混合物,5 μL预混液和3 μL无核酸酶的水。基因相对表达量使用2-△△Ct法进行计算,以GAPDH作为内参进行相对定量。引物序列见表3
1.2.6 CRISPR干扰实验:利用在线网站设计sgRNA序列(http://cistrome.org/SSC/)(表2)[6],将sgRNA构建到lentiGuide-Puro载体上,包装病毒,感染稳定表达dCas9-KRAB的细胞株[7]。48 h后加嘌呤霉素进行抗生素筛选,5 d后用Trizol法提取细胞总RNA,实时定量PCR检测基因表达。
1.3 统计学分析
1.3.1 差异表达基因的筛选:使用HISAT2、Stringtie、Ballgown套件流程分析RNA-seq的测序结果[8],差异表达基因定义为Qvalue < 0.05,且表达倍数差异不低于1.5倍的基因。
1.3.2 GO功能显著性富集分析:将差异表达基因向Gene Ontology数据库(http:www.geneontology.org/)的各个生物学过程条目映射,计算每个条目内的基因数目。
2 结果
2.1 rs6858698位点所在的DNA片段的调控活性
rs6858698-G DNA片段和rs6858698-C DNA片段均具有较强的增强子活性(P<0.05),其中rs6858698-C DNA片段所具有的增强子活性明显高于rs6858698-G DNA片段所具有的增强子活性,约有1.69倍(图1)。

表1 PCR引物序列Table 1 Primers used for PCR

表2 sgRNA靶点以及寡核苷酸序列Table 2 Oligos sequences of sgRNAs for knockin and CRISPRi

表3 RT-PCR引物序列Table 3 Primers used for RT-PCR

*P<0.001 comparing control group图1 双荧光素酶报告基因系统检测rs6858698位点的功能Fig 1 Dual-luciferase reporter assay was performed to detect the function of n=3)
2.2 CRISPR/Cas9系统介导的基因组编辑
野生型HEK293T细胞基因组DNA测序显示rs6858698位点的基因型为GG,利用CRISPR/Cas9系统进行基因组编辑后,成功将该位点的基因型由GG变为CC(图2)。
2.3 RNA-seq分析鉴别差异表达基因
根据差异表达倍数大于1.5和q值小于0.05,在野生型(rs6858698-G)和突变型(rs6858698-C)的HEK293T细胞中共筛选出140个差异表达基因。其中上调的差异表达基因有53个,下调的差异表达基因有87个。上调和下调的部分差异表达基因(表4,5)。GO生物学过程富集分析显示参与的生物学过程主要涉及蛋白质异二聚化活性、细胞黏附分子结合、 钙黏蛋白结合、 泛素样蛋白结合酶结合、泛素蛋白结合酶结合、组蛋白结合、染色质DNA结合、组蛋白去乙酰化酶结合、未折叠蛋白结合、核小体结合、核小体DNA结合、蛋白激酶活化剂活性和核定位序列结合(P<0.05)(图3)。
2.4 实时定量PCR验证差异表达基因的结果
选取了锌指蛋白595(zinc finger protein 595, ZNF595)、硒代半胱氨酸合成酶(selencysteine synthase,SEPSECS)、羰基还原酶(carbonyl reductase 4,CBR4)、甲基固醇单加氧酶1(methylsterol monooxygenase 1,MSMO1)和链非编码RNA 02506 (long non-coding RNA 02506,LINC02506)基因进行实时定量PCR检测,基因表达结果与RNA-seq分析结果一致,且均有显著性差异(P<0.05)(图4)。
2.5 rs6858698区域与差异表达基因之间的调控关系
利用CRISPR干扰实验(CRISPR interference,CRISPRi)抑制rs6858698区域的调控活性后,发现ZNF595、SEPSECS、CBR4、焦磷酸酶2(pyrophosphatase 2,PPA2)的基因表达量显著下调(P<0.01)(图5)。
3 讨论
本研究表明rs6858698位点具有等位基因特异性的调控活性。通过CRISPR系统所介导的基因组编辑,在293T细胞内定点突变了rs6858698位点的基因型。RNA-seq测序分析发现该位点不同等位基因所介导的调控活性差异,可以影响血管内皮生长因子A(vascular endothelial growth factor A,VEGFA)、转化生长因子1 (transforming growth factor beta 1, TGFB1)、细胞周期蛋白依赖激酶抑制因子1A(cyclin dependent kinase inhibitor 1A,CDKN1A)、原癌基因c-JUN(proto-oncogene c-Jun, JUN)、早期生长反应蛋白(early growth response protein2,EGR-2)等与慢性淋巴细胞白血病发生发展相关的基因的表达。
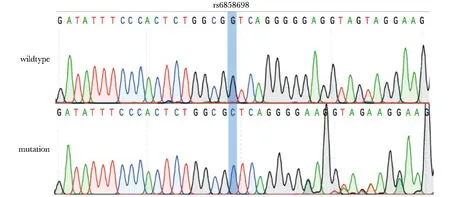
图2 rs6858698位点基因组编辑成功后的测序鉴定结果Fig 2 Sanger sequencing results of rs6858698-containing region in rs6858698-G and rs6858698-C cells

genefoldchangeP-valQ-valFGF135.540.00070.0261ELFN1-AS13.910.00050.0240OAZ23.840.00030.0196HSPA83.790.00120.0317MAGEA23.590.00330.0478DNAJB13.000.00060.0249FOXO42.400.00110.0302APOBEC3A2.240.00190.0365MSMO11.870.00160.0345CDKN1A1.770.00200.0379ZNF5951.750.00070.0268TGFB11.590.00150.0330CBX61.570.00130.0321

表5 差异表达下调部分已知基因列表Table 5 List of down-regulation part of the known genes
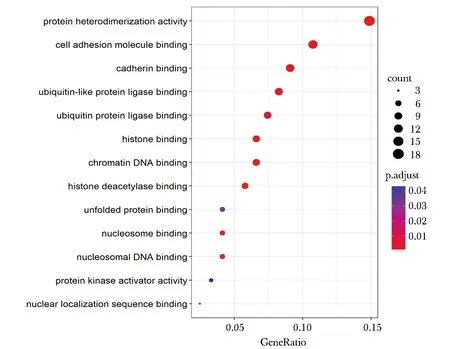
图3 GO富集分析结果Fig 3 GO enrichment analysis results
根据Oncomine网站的临床样本数据分析可知,VEGFA、JUN、CDKN1A、EGR2、CBR4、PPA2、SEPSECS等基因在慢性淋巴细胞白血病患者和正常人之间也存在显著的表达差异[9 -10]。其中VEGFA是一种重要的血管内皮生长因子,可以明显影响VEGF在细胞中的含量, 对慢性淋巴细胞白血病患者的预后具有指导意义[11]。所以,本研究中所分析出的差异表达基因对探究rs6858698位点影响慢性淋巴细胞白血病易感性的分子机制具有一定的指示作用。

Expression of SEPSECS, ZNF595, CBR4, MSMO1 and LINC02506 in rs6858698-C cells were down-regulated compared with rs6858698-G cells; *P<0.05, **P<0.01, ***P<0.001 compare with rs6858698-G group图4 实时定量PCR鉴定的结果Fig 4 Validation of gene expression changes by real-time quantitative PCR(RT-qPCR)
全基因组和全外显子组测序发现,慢性淋巴细胞白血病患者的基因组DNA内存在较少的基因突变及拷贝数变异,基因的异常表达主要是由于表观基因组异常所导致[12]。而在本实验中,通过GO功能富集分析显示,差异表达基因也主要富集在染色质构象、组蛋白修饰等与表观修饰相关的生物学过程。这也再次提示表观基因组异常在慢性淋巴细胞白血病发生发展中具有重要作用,也从一定程度上解释了rs6858698位点与慢性淋巴白血病发生风险之间的关联性。
综上所述,本实验通过构建定点突变的细胞株,分析了白血病易感位点rs6858698的不同基因型对基因表达的影响,初步鉴定了受到rs6858698位点调控的基因,为进一步研究rs6858698影响慢性淋巴细胞白血病易感性的分子机制提供了线索。
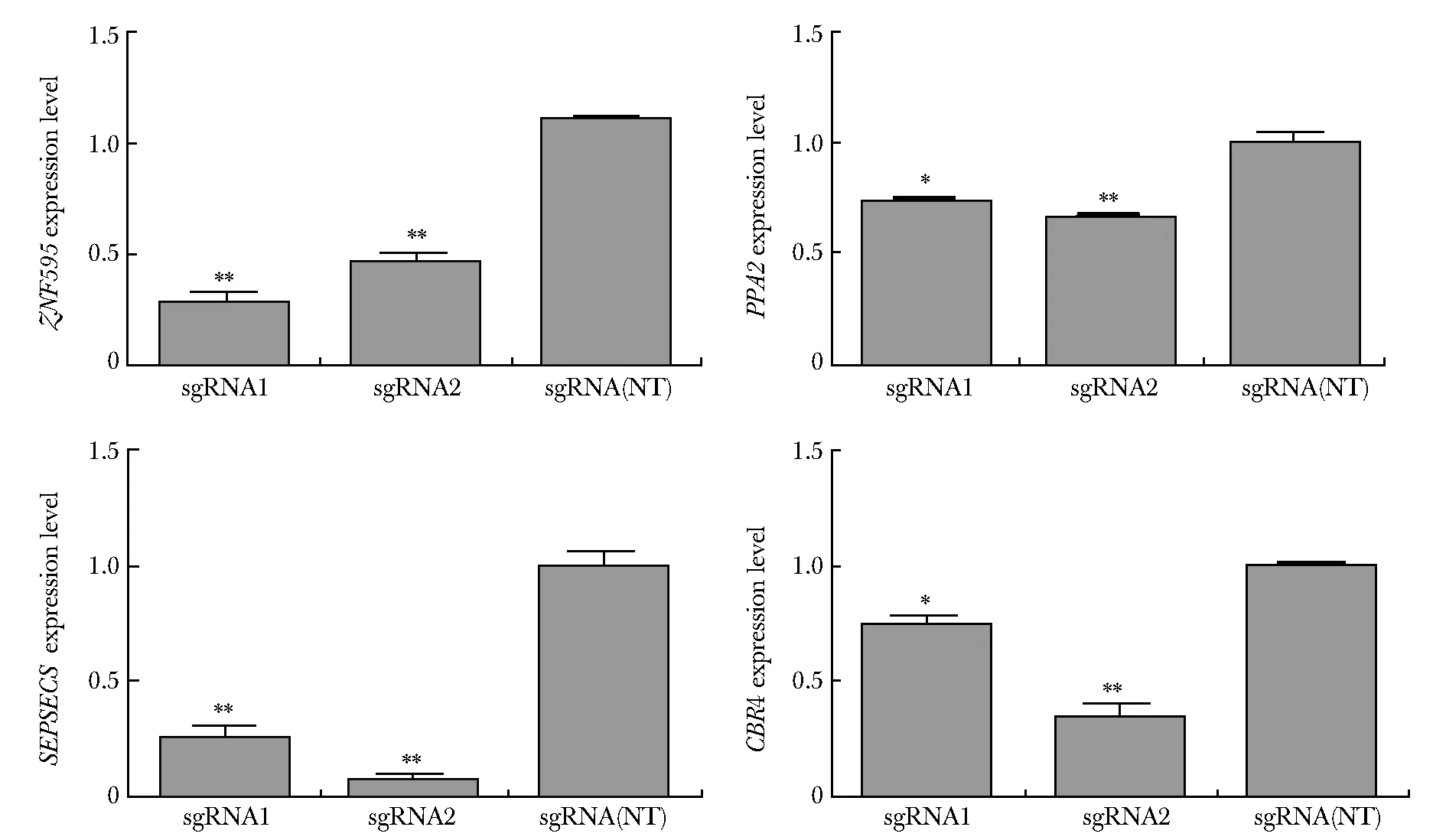
ZNF595,PPA2,SEPSECSandCBR4 in sgRNA1 and sgRNA2 groups were down-regulated compared with sgRNA(NT) group;*P<0.01,**P<0.001 compared with sgRNA(NT) group

参考文献:
[1] Welter D, MacArthur J, Morales J,etal. The NHGRI GWAS catalog, a curated resource of snp-trait associations [J]. Nucleic Acids Res, 2014, 42: D1001-D1006.
[2] Huang Q, Whitington T, Gao P,etal. A prostate cancer susceptibility allele at 6q22 increases rfx6 expression by modulating hoxb13 chromatin binding [J]. Nat Genet, 2014, 46: 126- 135.
[3] Speedy HE, Di Bernardo MC, Sava GP,etal. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia [J]. Nat Genet, 2014, 46: 56- 60.
[4] ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome [J]. Nature, 2012, 489: 57- 74.
[5] Richardson CD, Ray GJ, DeWitt MA,etal. Enhancing homology-directed genome editing by catalytically active and inactive crispr-Cas9 using asymmetric donor DNA [J]. Nat Biotechnol, 2016, 34: 339- 344.
[6] Xu H, Xiao T, Chen CH,etal. Sequence determinants of improved crispr sgrna design [J]. Genome Res, 2015, 25: 1147- 1157.
[7] Larson MH, Gilbert LA, Wang X,etal. Crispr interference (crispri) for sequence-specific control of gene expression [J]. Nat Protoc, 2013, 8: 2180- 2196.
[8] Pertea M, Kim D, Pertea GM,etal. Transcript-level expression analysis of RNA-seq experiments with hisat, stringtie and ballgown [J]. Nat Protoc, 2016, 11: 1650- 1667.
[9] Haferlach T, Kohlmann A, Wieczorek L,etal. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the international microarray innovations in leukemia study group [J]. J Clin Oncol, 2010, 28: 2529- 2537.
[10] Haslinger C, Schweifer N, Stilgenbauer S,etal. Microarray gene expression profiling of b-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and vh mutation status [J]. J Clin Oncol, 2004, 22: 3937- 3949.
[11] Lozano-Santos C, Martinez-Velasquez J, Fernandez-Cuevas B,etal. Vascular endothelial growth factor a (vegfa) gene polymorphisms have an impact on survival in a subgroup of indolent patients with chronic lymphocytic leukemia [J]. PLoS One, 2014, 9: e101063. doi: 10.1371/journal.pone.0101063
[12] Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia [J]. Nat Rev Cancer, 2016, 16: 145- 162.
