水分子对Cl在Fe(111)面吸附影响的密度泛函理论研究∗
2018-05-15郝义磊王洪博姜云瑛陈生辉李春玲孙霜青胡松青
石 鑫,郝义磊,王洪博,姜云瑛,陈生辉,李春玲,孙霜青,胡松青
(1.中石化西北油田分公司石油工程技术研究院,新疆乌鲁木齐830011;2.中国石油大学(华东)理学院,山东青岛266580)
0 引言
海洋蕴藏着丰富的矿产资源和油气资源,是人类未来的能源宝库.近年来,我国把海洋资源的开发和利用提升为国家战略,并加速了海洋基础设施和工业设施的建设.然而,船舶、钻井平台等海上作业设施在海水环境中会发生严重的腐蚀,成为海洋经济发展的重大阻碍,因此,开展海洋腐蚀研究具有突出的现实意义[1].
海水中含有大量Cl,溶解于水中的Cl对金属腐蚀有重要影响[2].文献[3]表明:Cl−半径较小,穿透、吸附能力强,溶解于水有利于其实现扩散与迁移;另外,Cl−溶于水为腐蚀原电池提供电解质溶液,是金属发生电化学腐蚀的前提.水分子与Cl−的共同作用使金属表面发生孔蚀[4],在生产生活中造成极大危害.铁是工业生产中使用最广泛的金属材料,铁表面的吸附问题一直是国内外科研工作者研究的热点问题,人们在实验和理论方面做了大量的研究工作.Sh.Hassani等通过实验发现:溶液中的Cl−在铁表面吸附能够加速铁的腐蚀.铁的腐蚀速率随Cl−浓度的升高(0∼26%)呈现先增加后减小的趋势,在Cl−的浓度为3%(海水中Cl−的浓度)时,铁的腐蚀速率达到最大值[5].Qu等[6]的研究发现:A3钢在含SO2环境中发生腐蚀时,NaCl不仅会加速其初期腐蚀速度,还会造成腐蚀产物表面开裂,发生局部腐蚀.在模拟研究中,Freitas等[7]采用密度泛函理论研究了H2O分子在Fe(100)面的吸附和解离行为,发现H2O分子和OH−在Fe表面的吸附方式分别为物理吸附和化学吸附,H2O分子在Fe表面的解离势垒在0.94−1.04 eV之间.Das等[8]则通过分子动力学方法模拟了Fe表面的氧化过程,研究发现H2O分子解离出的H+会优先渗入Fe表面,接受电子并充当O2的载体,导致金属氧化并减弱金属键强度.总之,铁材料在含氯环境中的点蚀机理已形成较为完善的实验研究体系,人们在实验规律的基础上获得腐蚀机理,却无法从电子层次阐释金属与腐蚀粒子间的相互作用.量子化学计算可以在电子层次揭示化学反应的本质,在研究Cl腐蚀机理方面有着独特的优势,却鲜有人尝试.本文采用密度泛函方法通过计算几何结构、吸附能、键性质和电子特性,研究了水分子的引入对Cl在Fe(111)面吸附性质产生的影响.
1 计算方法与模型
1.1 计算方法
本文的所有计算均采用CASTEP(Cambridge Sequential Total Energy Package)[9]软件包完成,该软件采用基于密度泛函理论的平面波赝势方法.其中,电子交换关联相互作用采用广义梯度近似(GGA)中的Perdew-Wang PW91[10]交换关联函数描述,波函数采用Broyden-Fietcher-Goldfarb-Shanno(BFGS)方法优化,离子实与价电子之间相互作用采用超软赝势(Ultrasoft Pseudopotential)描述,原子总能量、原子受力、最大张力和位移公差的收敛判据分别为2.0×10−5eV/atom、0.05 eV/˚A、0.1 GPa、0.005˚A,截断能为400 eV,自洽计算收敛精度为2.0×10−6eV/atom.能量计算都在倒易空间中进行,釆用周期性边界条件.吸附模型的布里渊区k点取样为4×4×1,Fe晶胞、Cl原子和H2O分子优化时布里渊区k点取样为12×12×12.由于Fe体系具有磁性,因此计算考虑自旋极化.
1.2 计算模型
本文的吸附模型由Fe(111)面吸附基底和Cl、H2O吸附粒子构成.Fe(111)表面采用7层p(2×2)的Fe原子平板模型模拟(见图1(a)),相邻平板模型之间添加厚度为15˚A的真空层,以保证相邻层间的相互作用足够小.文献[11]报导大多数过渡金属表面1∼3层原子容易发生驰豫,4层以下原子则不会发生明显的弛豫现象.因此固定下面4层原子作为体相原子,上面3层原子作为表面原子自由移动.根据吸附粒子质心在金属表面的投影位置,Fe(111)表面的吸附位点可分为Top位(T位),Bridge位(B位),HCP Hollow位(H位)以及FCC Hollow位(F位)四种吸附位点[12,13](见图1(b)).构建吸附模型时,将Cl先后放置在四种吸附位点的上方,研究Cl在四种位点的吸附特性.然后,选取其中较为稳定的两种吸附构型,将H2O分子放置在Cl周围,研究H2O分子对Cl在Fe(111)面吸附的影响.
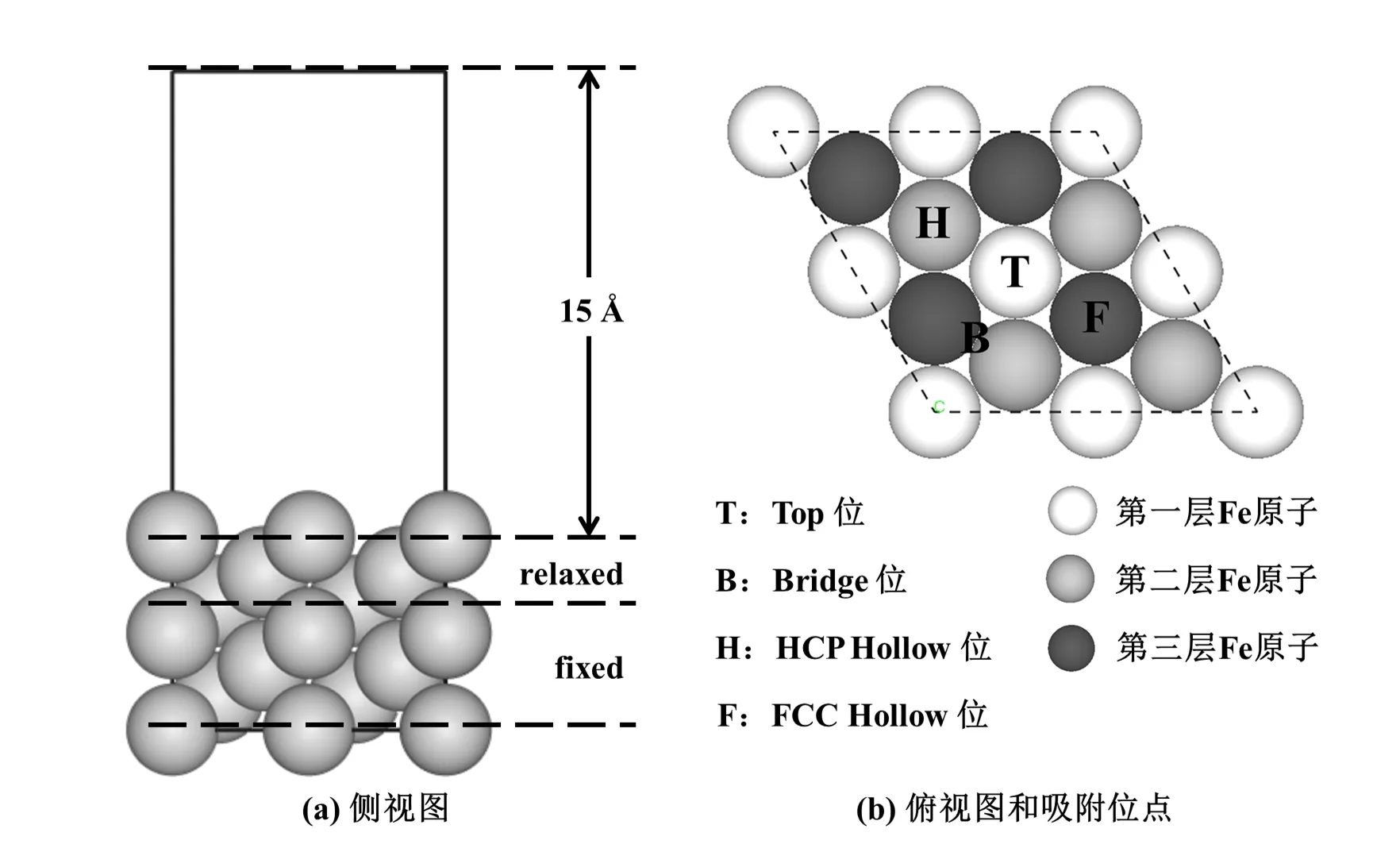
图1 Fe(111)表面的周期型模型及表面吸附位点
1.3 模型验证
为进一步验证计算参数和模型的精度与可靠性,本文还计算了Fe的晶胞参数,H2O分子的结构参数以及Fe(111)面的表面能及功函数,并将其与实验值进行比较.表面能和功函数的定义分别如式(1)和式(2)所示.计算结果如表1所示,优化后Fe晶格常数值为2.83˚A,与实验值2.87˚A相差1.4%;优化后H2O的键长为0.98˚A,键角为104.6˚,与实验值0.97˚A和104.5˚相差1%和0.1%;Fe(111)面模型的表面能和功函数分别为2.43 J/m2和4.13 eV,这与实验值2.41 J/m2和4.47 eV相差0.8%和7.6%,这说明所建立的模型是可靠的,能够满足计算精度的需求.

其中,Esurf为表面能,A为平板模型的表面积,Eslab为模型总能量,Ebulb为体材料中的原胞能量,n为模型中包含的原子数.

其中,φ为表面功函数,Evacuum为真空能级,Efermi为费米能.

表1 计算参数与实验参数对比
2 结果与讨论
2.1 稳定吸附构型
图2所示为Cl在Fe(111)面T位、B位、H位和F位4种吸附位点优化前后的吸附构型.Cl单独在Fe(111)面吸附后,T位和F位吸附的Cl最终稳定于T位附近,而B位和H位吸附的Cl最终稳定在H位附近,说明T位和H位是Cl在Fe(111)面吸附的两个相对稳定位点.此外,为了研究H2O分子对Cl吸附构型的影响,本文在Cl的两种稳定吸附构型(T位和H位)中分别引入一个H2O分子,计算获得了Cl和H2O分子共吸附稳定构型(如图4所示).在H2O分子的作用下,T位Cl吸附位置无变化,而H位Cl的吸附位置出现偏移,这说明H2O分子对H位吸附的Cl产生了明显影响.
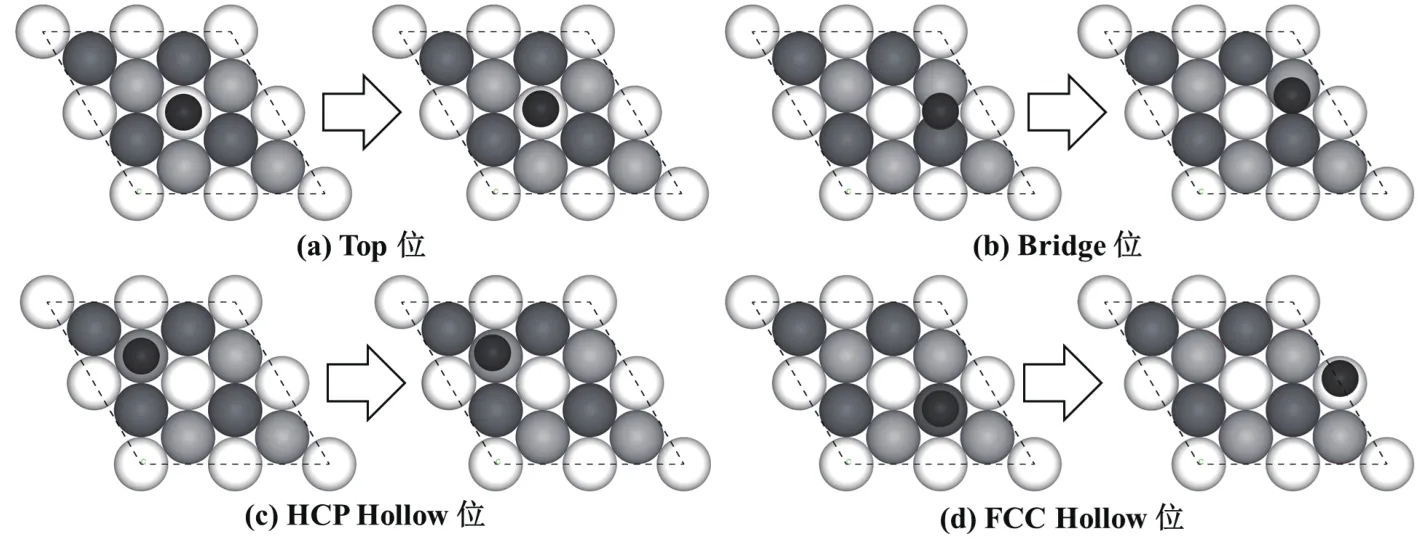
图2 Cl在Fe(111)面的吸附
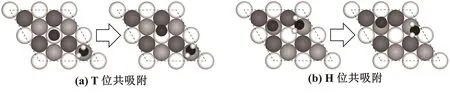
图3 Cl和H2O分子在Fe(111)面的共吸附
2.2 水分子对Cl-Fe相互作用的影响
为了研究引入H2O分子后,对Cl-Fe之间相互作用产生的影响,本文还计算了Cl的吸附能、Cl与Fe(111)表面距离及Cl-Fe键布局,见表2.
吸附能是指吸附质在表面稳定吸附后释放出的能量,其计算公式[17−19]如式(3)所示
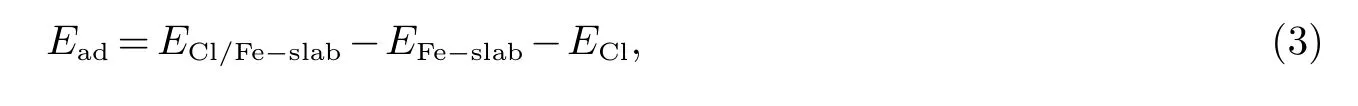
式中,Ead为Cl在Fe(111)面的吸附能,ECl/Fe−slab为优化后Cl-Fe体系的总能量,EFe−slab为Fe表面的能量,ECl为Cl的能量.如表2,所有模型的吸附能Ead均为负值,说明吸附反应放热,可以自发产生.Cl单独在T位吸附时,吸附能为-4.64 eV,Cl-Fe距离为2.170˚A,Cl单独在F位吸附时,吸附能为-4.59 eV,Cl-Fe距离为2.185 ˚A,这两种模型的结构参数较为接近;Cl单独在B位吸附时,吸附能为-4.81 eV,Cl-Fe距离为1.639˚A,Cl单独在H位吸附时,吸附能为-4.89 eV,Cl-Fe距离为1.620˚A,这两种模型的结构较为接近;Cl吸附模型中引入H2O分子后,T位吸附的吸附能由-4.64 eV变为-5.39 eV,H位吸附的吸附能由-4.89 eV变为-5.58 eV.由于吸附能的数值越负吸附越稳定,说明H2O分子可以促进Cl在Fe(111)面的吸附.键布局是指电子在化学键上的分布,它能够反映化学键的强度和类型[20],负值代表反键,0代表离子键,正值代表共价键,且绝对值越大键的强度越大.由表2可知,Cl在T位吸附时,Cl仅与其下方的Fe原子成键,与表面的其它Fe原子相互作用较小,而Cl在H位吸附后,Cl可以同它下方的三个Fe原子同时成键,虽然每个键的强度和共价性不及T位形成的单键,但这三个键的共同作用却强于T位吸附形成的单键,这就导致了Cl在H位的吸附能比T位吸附更负.引入H2O分子后,T位吸附Cl与Fe(111)面的距离dCl−Fe由2.170 ˚A减小为2.154 ˚A,Cl-Fe键布局由0.81增大到0.82,键强和共价性略有提高;H位吸附Cl与Fe(111)面的距离dCl−Fe由1.620˚A减小为1.596 ˚A,Cl-Fe键布局由0.39、0.20、0.16变为0.43、0.33,键强和共价性显著提高.
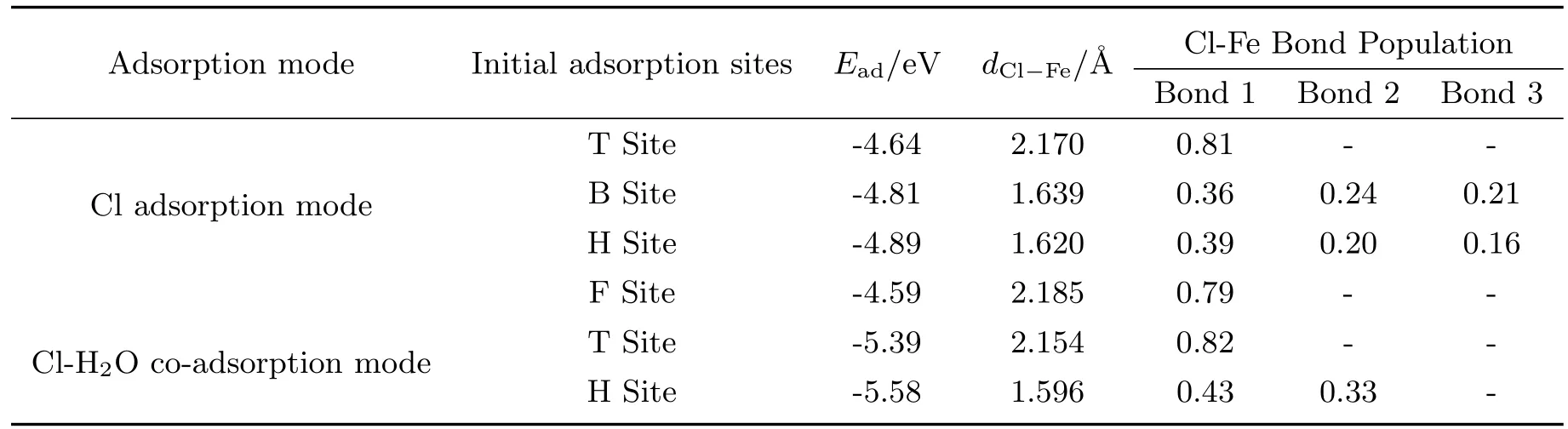
表2 不同吸附构型的吸附能和成键参数
由图3(b)和表2可知,在H2O分子作用下,Cl在H位的吸附模型发生改变,这种变化主要体现在Cl吸附位置偏移,Cl与Fe成键数减少以及Cl-Fe键布局增大.原因可能是引入H2O分子后,H2O分子中的O2−由于电负性较强与Cl竞争吸附位点,对T位和H位吸附模型产生了不同影响效果.当Cl在T位吸附时,成键强度较大,重叠布局为0.82,Cl-Fe键不容易被破坏;当Cl在H位吸附时,虽然Cl-Fe成键数量较多,但每个键的重叠布局都小于0.5,这种结构容易在H2O分子影响下发生转变,从而导致Cl的吸附位点发生偏移.
2.3 Cl-H2O吸附对Fe(111)面的影响
H2O分子在影响Cl吸附于Fe(111)面的过程中,也会对Fe表面本身产生一定的影响.表3列出了不同吸附结构中Fe(111)面的层间距、功函数以及表层Fe原子的平均电荷数.由表3可知,表层Fe原子的平均电荷数全部为正值,表明金属表层的原子有氧化趋势.Cl在Fe表面吸附后,表层Fe原子的平均电荷数由0.25增至0.30左右,变化大于16%,这表明Cl在Fe表面的吸附能够加速Fe表面的氧化.在此基础上再引入H2O分子,Fe表面的电荷数几乎不变,表明H2O分子不具有加速氧化作用.表面的层间距能反映表面的稳定性,层间距越大,表面稳定性越差.由表3可知,Cl单独在Fe(111)面吸附对Fe的层间距影响很小,其变化率小于2%.但引入H2O分子后,T位吸附模型的层间距Δd1−2增大了28%,H位吸附模型层间距Δd1−2增大了13%,这表明H2O分子能促进Fe(111)面表层原子剥离,从而影响Fe基体的稳定性,Cl则不具备这种作用.为了验证Fe表面在Cl和H2O同时存在时具有较强的腐蚀敏感性,本文还计算了模型的表面功函数,功函数的值越小,表明体系的化学稳定性越差.如表3所示,Cl单独吸附后,表面的功函数略有减小,其变化率小于3%,而引入H2O后表面的功函数明显减小,其变化率大于14%.这说明Cl和H2O分子的协同作用确实是影响Fe表面稳定性的主要因素.
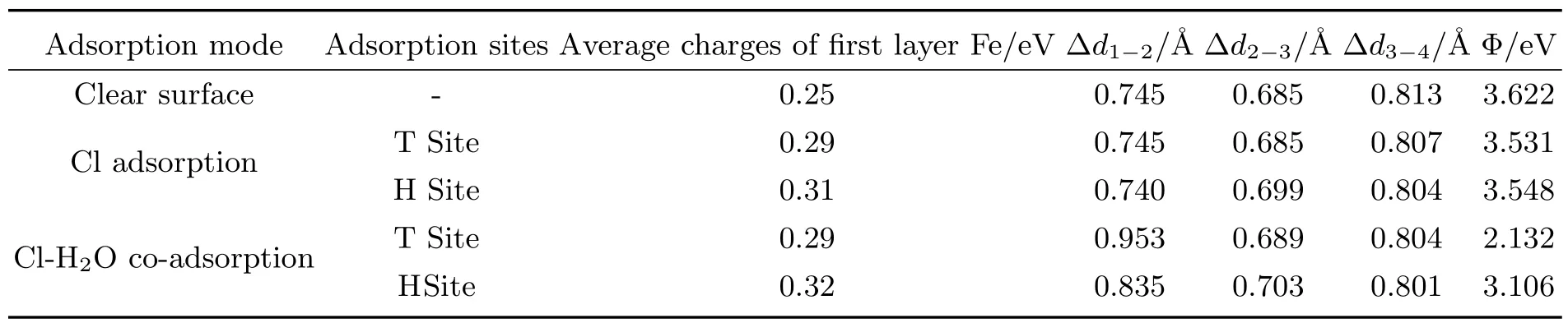
表3 不同吸附构型的层间距和电子性质
2.4 PDOS分析
PDOS(分波态密度)能够描绘某个原子轨道上电子对态密度的贡献,使之成为细致分析原子之间成键性质的有效工具[21].本文计算了T位和H位两种吸附模型的PDOS图(如图4所示).(a)图和(d)图分别为吸附前T位和H位Cl、H2O、Fe的PDOS图,(b)图和(e)图分别为Cl单独吸附在T位和H位时Cl和Fe的PDOS图, (c)图和(f)图分别为Cl和H2O在T位和H位共吸附时Cl、H2O、Fe的PDOS图.对比吸附前后的PDOS图,无论是T位吸附还是H位吸附,吸附后Cl的3s和3p轨道、O的2s和2p轨道以及H的1s轨道峰都发生左移,但H位吸附的左移程度大于T位吸附.这说明吸附发生后,Cl、O、H的能量都有所降低,而H位吸附时能量降低的更多.由(b)和(e)图可知,Cl在Fe(111)面吸附后,Cl的3p轨道电子峰有拓宽趋势,且与Fe的3d轨道电子峰有重叠,这说明Cl的3p轨道与Fe的3d轨道之间存在明显的杂化耦合作用.在(e)、(f)两图中,Fe的3d轨道与Cl的3p轨道重合程度明显不如(b)、(c)图,说明H位吸附的Cl-Fe相互作用明显小于T位吸附.由(b)图到(c)图,Cl的3p轨道和Fe的3d轨道峰型没有明显变化,而由(e)图到(f)图时它们发生了明显的变化,说明H位在引入H2O后,Cl-Fe成键性质发生了明显的改变.
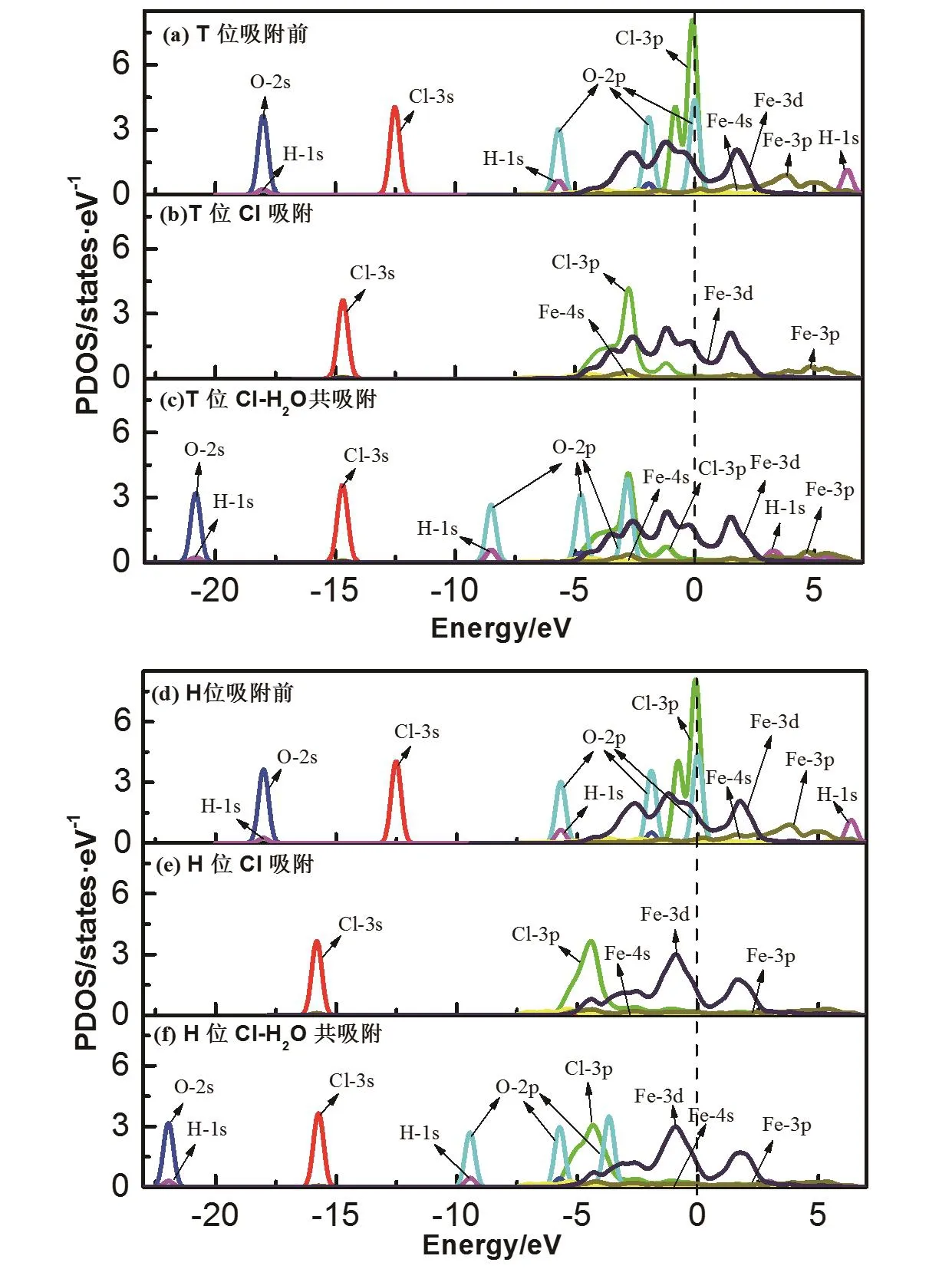
图4 T位吸附和H位吸附的PDOS图
3 结论
本文采用密度泛函理论研究了Cl在Fe(111)表面的单独吸附构型以及Cl-H2O共吸附构型,通过吸附能、成键参数、电荷分布和分波态密度等参量分析了H2O分子对Cl在Fe表面稳定吸附构型的影响.结果表明:
(1)Cl能够自发稳定吸附于Fe(111)表面的T位和H位,且在H位的吸附能更大;Cl不能在B位和F位稳定吸附,其吸附构型转化为T位和H为吸附.
(2)引入H2O分子后,H2O分子会促进Cl在Fe(111)面的吸附能增加,但同时也会影响吸附构型的稳定性,T位吸附的Cl因存在较强的Cl-Fe键而不易产生结构改变,H位吸附的Cl则会由于结构原因发生吸附位点的偏移并伴随Cl-Fe键性质的改变.
(3)Cl单独在Fe(111)面吸附时,会导致表层Fe原子的平均正电荷数增加;H2O分子引入后,则导致第一层Fe原子与第二层Fe原子之间的距离明显增大.Cl和H2O分子的协同作用是导致Fe表面化学稳定性降低的原因,在共吸附过程中伴随了Fe表面功函数的明显降低.
(4)PDOS分析表明:吸附过程发生时,吸附粒子的能量均有所降低;Cl与Fe表面的成键方式为Cl的3p轨道与Fe原子的3d轨道发生杂化耦合;H2O分子的引入会使H位吸附的Cl-Fe成键性质改变,T位吸附模型的Cl-Fe键则没有改变.
参考文献:
[1]Han E H,Chen J M,Su Y J,et al.Corrosion protection techniques of marine engineering structure and ship equipment——current status and future trend[J].Materials China,2014,33(2):65-76.
[2]郭良生.水溶液中Cl−对钢铁腐蚀的影响及其对策[J].表面技术,1998,27(4):23-24.
[3]刘薇,王佳.海洋浪溅区环境对材料腐蚀行为影响的研究进展[J].中国腐蚀与防护学报,2010,30(6):504-512.
[4]Liu J C,Park S W,Nagao S,et al.The role of Zn precipitates and Cl−anions in pitting corrosion of Sn–Zn solder alloys[J].Corrosion Science,2015,92:263-271.
[5]Hassani S,Roberts K P,Shirazi S A,et al.Flow loop study of NaCl concentration eあect on erosion,corrosion,and erosioncorrosion of carbon steel in CO2-saturated systems[J].Corrosion,The Journal of Science and Engineering,2012,68(2):026001-1-026001-9.
[6]Qing Q U,Yan C,Wei B,et al.Inf l uence of NaCl deposition on atmospheric corrosion of zinc in the presence of SO2[J].Acta Metallrugica Sinica,2001,18(5):552-555.
[7]Freitas R,Rivelino R,De B M,et al.Dissociative adsorption and aggregation of water on the Fe(100)surface:a DFT study[J].The Journal of Physical Chemistry C,2012,116(38):20306-20314.
[8]Das N,Suzuki K,Takeda Y,et al.Quantum chemical molecular dynamics study of stress corrosion cracking behavior for fcc Fe and Fe–Cr surfaces[J].Corrosion Science,2008,50(6):1701-1706.
[9]Delley B.An all-electron numerical method for solving the local density functional for polyatomic molecules[J].The Journal of chemical physics,1990,92(1):508-517.
[10]Perdew J P,Ziesche P,Eschrig H.Electronic structure of solids’91[M].Berlin:Akademie Verlag,1991.
[11]吴越.催化化学[M].北京:科学出版社,2000:675.
[12]张凤春,李春福,孙延安,等.硫在Fe(111)面吸附的密度泛函研究[J].原子与分子物理学报,2013,30(002):328-336.
[13]Zhang F C,Li C F,Sun Y A,et al.Density functional theory study on sulfur adsorption on Fe(111)surface[J].J Atom Mol Phys,2013,30(002):328-336.
[14]赵巍,汪家道,刘峰斌,等.H2O分子在Fe(100),Fe(110),Fe(111)表面吸附的第一性原理研究[J].物理学报,2009(5):3352-3358.
[15]Blonski P,Kiejna A.Structural,electronic,and magnetic properties of bcc iron surfaces[J].Surface science,2007,601(1):123-133.
[16]Eastman D.Photoelectric Work Functions of Transition,Rare-Earth,and Noble Metals[J].Physical Review B Condensed Matter,1970,2(1):1-2.
[17]Sorescu D.Adsorption and activation of CO coadsorbed with K on Fe(100)surface:A plane-wave DFT study[J].Surface Science,2011,605(3):401-414.
[18]Rochana P,Wilcox J.A theoretical study of CO adsorption on FeCo(100)and the eあect of alloying[J].Surface Science,2011,605(7):681-688.
[19]张福兰.CHx(x=2∼4)在Fe(110)表面吸附的DFT研究[J].原子与分子物理学报,2010(5):986-992.
[20]薛卫东,陈召勇,杨春,等.四方相BaTiO3铁电性的第一性原理研究[J].物理学报,2005,54(2):857-862.
[21]赵巍,汪家道,刘峰斌,等.Cl与H2O在Fe(100)表面共吸附的稳定结构与电子特性[J].科学通报,2009(6):740-746.
