耐热型高温热膨胀微球的制备及其发泡行为研究
2018-05-11,,,,,,
,, , ,,,
(1.浙江理工大学先进纺织材料与制备技术教育部重点实验室,杭州 310018; 2.浙江精通科技股份有限公司研发部,杭州 311121)
0 引 言
由低沸点烃类物质和聚合物壳壁构成的热膨胀微球在加热时体积能膨胀数十倍甚至上百倍[1]。热膨胀微球在膨胀前后能产生显著体积变化的特点,使其在材料的轻量化、图案化、立体化等方面有重要的应用价值[2-5]。近年来,对聚氯乙烯等塑料轻量化处理的热膨胀微球的需求逐年增加,对于此类应用,一般要求微球需同时具备高的膨胀倍率以及优良的高温稳泡性能。
目前,热膨胀微球的制备方法已有较多的国内外专利[6-10]和文献报道[11-13],主要是以低沸点的烷烃为发泡剂,丙烯腈为主单体,通过悬浮聚合法来制备。热膨胀微球的发泡能力受发泡剂组成、壳壁聚合物组成、发泡剂与壳壁聚合物的比例、微球的尺寸及其分布等众多因素的影响,因此针对微球发泡性能的调控较为复杂[14-17]。而且高的膨胀倍率和优良的高温稳泡性能往往难以兼顾,因此制备耐热型高温热膨胀微球具有较大的难度。丁婉等[18]研究了高温物理发泡微胶囊的制备及影响发泡性能的因素;Jonsson等[19]研究了交联剂对热膨胀微球在高发泡温度下膨胀特性的影响;朱效杰等[20]研究了含甲基丙烯酸的高温发泡微球的合成及其发泡性能。上述研究推动了高性能高温热膨胀微球的开发,但大部分研究只侧重对热膨胀微球的发泡温度和膨胀性能的研究,对膨胀后微球的高温稳泡性能研究较少。
本文在前人的研究基础上,通过对壳壁的合理设计,采用悬浮聚合方法,制备兼具高膨胀性能和优良高温稳泡性能的热膨胀微球。采用多种表征手段系统研究交联剂类型和用量这两个关键因素对微球形貌、发泡性能的影响规律,确定最佳的交联剂类型和用量。
1 实验部分
1.1 主要试剂和原料
丙烯腈(AN):CP,浙江精通科技股份有限公司;N,N-二甲基丙烯酰胺(DMAA):CP,浙江精通科技股份有限公司;甲基丙烯酸(MAA)、甲基丙烯酸甲酯(MMA):AR,天津市科密欧化学试剂有限公司;过氧化苯甲酰(BPO)、高分子型交联剂(P1)、二乙烯基苯(DVB)、三羟甲基丙烷三丙烯酸酯(TMPTA)、异戊烷、十二烷基硫酸钠(SDS):AR,阿拉丁试剂;异辛烷:AR,上海展云化工有限公司;氢氧化钠:AR,无锡市展望化工试剂有限公司;氯化镁:AR,天津市致远化学试剂有限公司;氯化钠:AR,天津市百世化工有限公司。
1.2 主要仪器和设备
高剪切分散乳化机:FA25,上海弗鲁克仪器有限公司;激光粒度分析仪:Mastersizer 2000,英国Malvern公司;扫描电镜(SEM):JSM-5610LV,日本电子(JEOL);光学显微镜:上海舜宇恒平科学仪器有限公司。
1.3 热膨胀微球的制备
将一定量氢氧化钠和氯化钠溶于去离子水中,得到1号水溶液。将一定量六水合氯化镁、SDS和亚硝酸钠溶于去离子水中,得到2号水溶液。用蠕动泵将1号水溶液缓缓滴入2号水溶液中,边滴边搅拌。1号水溶液滴完后,将分散液的pH值调至碱性,作为悬浮聚合体系的水相。按比例将AN、MMA、MAA、DMAA和交联剂混合,形成单体溶液,再将一定量的BPO溶于上述单体溶液中,最后再加入一定量发泡剂异辛烷和异戊烷,混合均匀后得到悬浮聚合体系的油相。将上述水分散液与油相混合,高速剪切分散,制得单体悬浮液。将制得的悬浮液倒入高压反应釜中,密封后加压。当温度达到预设温度后,开始计时,恒温反应20 h后,经冷却、卸压、出料、洗涤、过滤、干燥、筛选等工序后,制得干燥的热膨胀微球。
1.4 热膨胀微球粒径及其分布的测定
取1~2 g热膨胀微球样品,加到20~30 mL去离子水中,超声分散均匀后,用激光粒度仪测定热膨胀微球的平均粒径。粒径分布指数按公式(D90-D10)/D50计算,其中D90、D50和D10分别表示一个样品的累积粒径分布百分数达到90%、50%和10%时所对应的粒径,它们的物理意义表示尺寸小于该粒径的颗粒数占总颗粒数的量分别为90%、50%和10%。
1.5 热膨胀微球形貌观察
用光学显微镜观察颗粒形态以及用SEM观察热膨胀微球表面形貌和切片样品的形貌。切片样品的制备方法如下:将待测样品置于环氧树脂中进行包埋固化,液氮淬冷,然后在液氮环境下进行冷冻切片。
1.6 微球发泡性能的测试
称取0.1 g干燥的微球样品,置于25 mL的试管中,将装有试样的试管放入不同预热温度的烘箱中,待微球充分发泡后,记录膨胀后微球的宏观体积,得到微球发泡温度与膨胀微球宏观体积的曲线,将微球达到最大体积的最小发泡温度定为该样品的最佳发泡温度TF。用光学显微镜观察发泡前后的微球样品,并拍摄照片,分别统计200个发泡前后微球的粒径,计算得到相应的数均粒径do和d,并将两者的比值(d/do)定为微球的发泡倍率。将样品置于185 ℃的烘箱中,记录微球开始发泡至出现收缩的时间,将其定义为微球的稳泡时间,用于表征微球在高温下的热稳定性。
2 结果与讨论
2.1 热膨胀微球的颗粒特征
2.1.1 热膨胀微球的粒径及其分布
用激光粒度仪测定了悬浮聚合法制备的热膨胀微球的粒径及其分布,结果见图1。由图1可知,微球的粒径主要分布在20~250 μm范围内,其D10、D50和D90粒径分别为32.2、67.4 μm和148.9 μm。根据公式(D90-D10)/D50计算得到热膨胀微球的粒径多分散指数约为1.7。此外,图1显示在小于10 μm的区域内有一个小肩峰,说明样品中还存在少量小颗粒;大于250 μm区域中也出现了一个小肩峰,这可能是因为少量干燥后的微球在再分散处理过程中没有被完全打散所致。
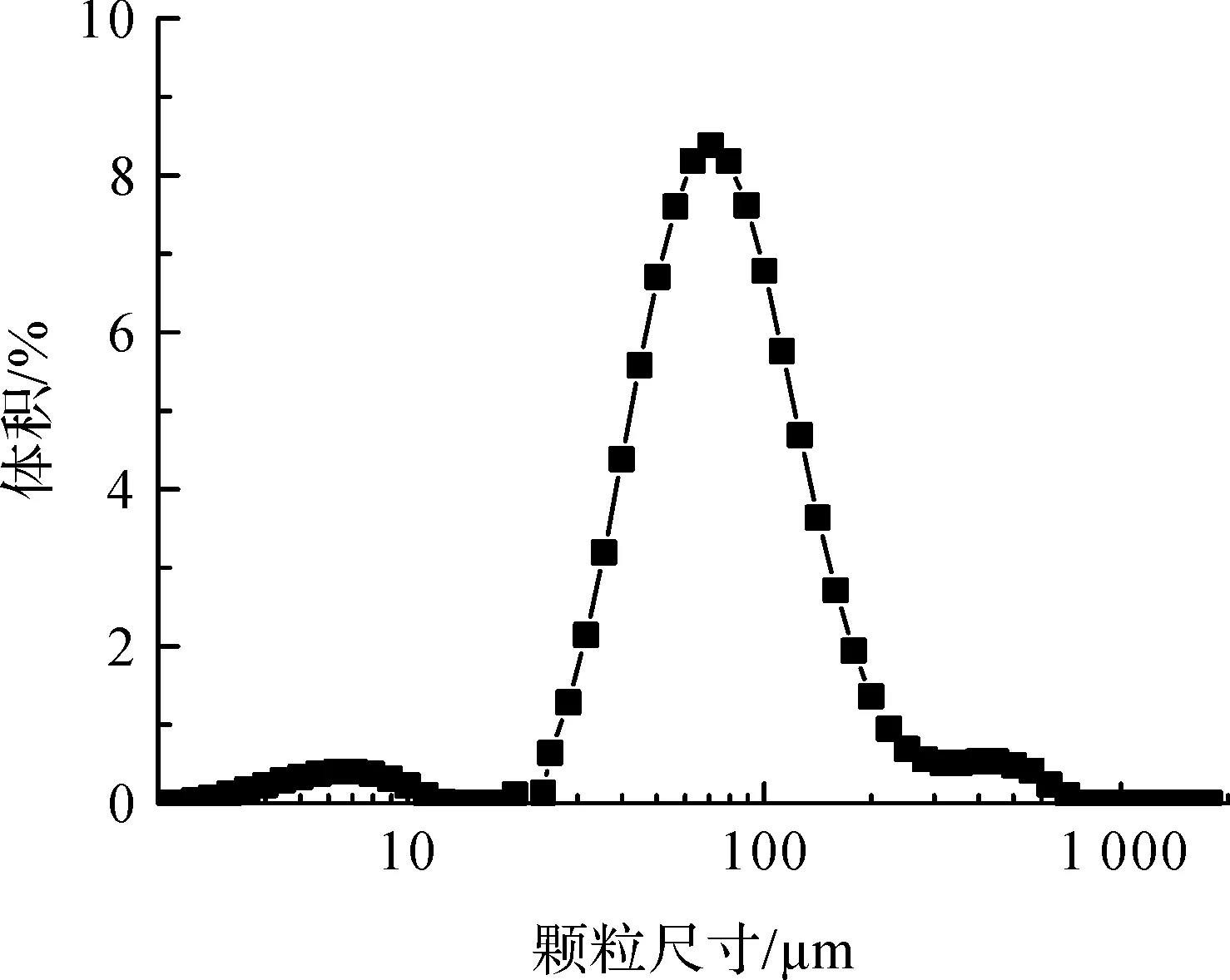
图1 热膨胀微球的粒径分布曲线
2.1.2 热膨胀微球的形貌
图2(a)是热膨胀微球的光学显微镜照片,由图可知,微球为非规则的球体,大部分微球的尺寸在60~70 μm范围内,此外,样品中还出现了少量小尺寸的颗粒,其尺寸为8~9 μm。上述结果与粒径测试结果相一致。用SEM进一步研究了热膨胀微球的形貌特征,结果见图2(b)。由图2(b)可知,热膨胀微球的壳壁出现了大量的凹陷和褶皱,这主要是由聚合物壳壁的机械强度和微球内外压强差引起的。当聚合反应结束后,反应体系在一定压力下被冷却至室温,被包覆在微球内的烷烃体积收缩,微球内压力下降,微球内外产生压差,驱动微球收缩。但须指出的是,微球并未发生均匀的收缩,形成尺寸较小的球体,而是出现了不均匀的收缩,形成了表面凹陷褶皱的颗粒。这是因为热膨胀微球的壳壁主要由丙烯腈、甲基丙烯酸甲酯和能形成氢键的极性单体单元构成,在室温至反应温度的温度范围内,壳壁聚合物部分结晶,且分子间作用力大,因此具有较大的刚性,与弹性球体均匀收缩不同,发生了不均匀的收缩,形成了不规则的球体。图2(c)是热膨胀微球切片后的SEM图,由图可知,热膨胀微球内有一明显的内腔,微球的聚合物壳壁厚度在3~4 μm之间。综合上述,通过悬浮聚合法成功合成了具有核-壳结构且壳厚度均匀的热膨胀微球。

图2 热膨胀微球的微观形貌图
2.2 热膨胀微球的发泡行为
热膨胀微球的发泡行为如图3所示。由发泡温度与膨胀后微球宏观体积的关系曲线(图3(a))可知,所制微球的起始发泡温度约为140 ℃,在155~165 ℃范围内,膨胀后微球的体积显著增加,当发泡温度达到185 ℃后,微球的膨胀体积基本保持不变,说明此时微球已达到最大膨胀状态,故将185 ℃定为该微球的最佳发泡温度TF。
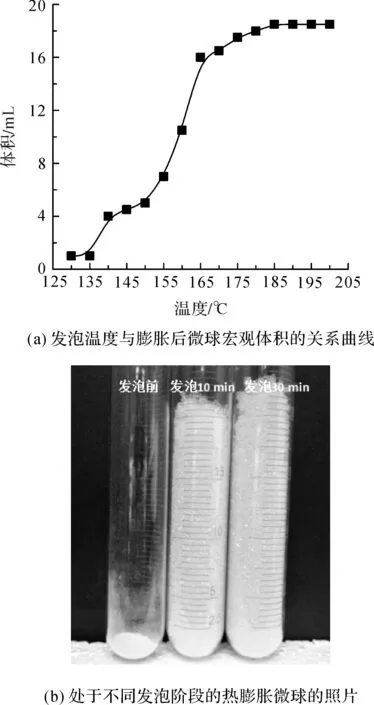
图3 热膨胀微球的发泡行为
为更定量地表征热膨胀微球的发泡性能,根据微球显微镜图片统计得到了微球发泡前后的数均粒径,并将两者的比值定为微球的发泡倍率。热膨胀微球的光学显微镜图见图4,由图可知,发泡前微球颗粒的数均粒径为(68±13) μm,微球在185 ℃条件下发泡后,数均粒径显著增加到了(300±120) μm,可得微球的发泡倍率约为4.4倍。
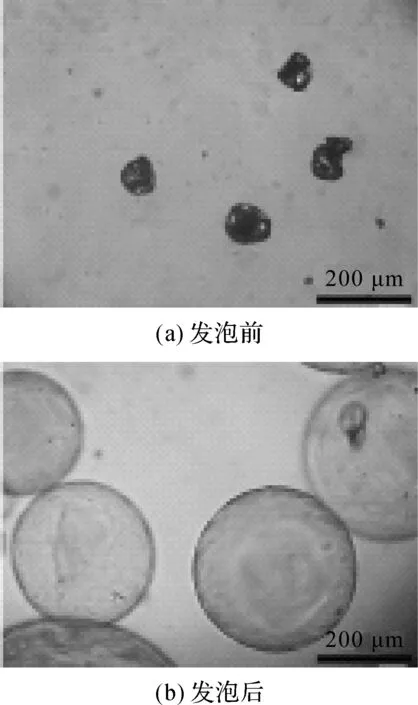
图4 热膨胀微球的光学显微镜图
对于高温型热膨胀微球,其在发泡后的高温稳泡性能亦是决定其使用性能的重要指标。结合图3可知,在185 ℃的温度条件下,在10 min内微球的膨胀过程基本结束,将发泡时间延长至30 min,膨胀后的微球只出现了轻微的收缩。综上所述,所制微球的TF为185 ℃,在该发泡温度下微球具有优良的膨胀性能和高温稳泡性能。
2.3 交联剂对热膨胀微球发泡行为的影响
2.3.1 交联剂类型
在保持其他反应参数不变的条件下,考察了交联剂类型对热膨胀微球发泡行为的影响。由图2(b)和5(a)-(c)可知,不同交联剂类型条件下合成的微球均为非规则的球体,表面存在较多的凹陷,交联剂类型对热膨胀微球的形貌影响较小。由图4(b)和5(d)-(f)可知,在特定的发泡温度下,这些微球都能受热膨胀,但其发泡倍率与交联剂类型密切相关。

图5 不同交联剂类型条件下制备的热膨胀微球发泡前的SEM图(a-c)及热膨胀微球发泡后的光学显微镜照片(d-f)
表1列出了交联剂类型对TF、发泡倍率和稳泡时间等发泡性能的影响。由表1可知,以TMPTA为交联剂的微球发泡倍率仅为2.9倍,明显小于其他交联剂条件下制得的微球。此外,在微球膨胀实验过程中还发现以TMPTA为交联剂的微球出现了较多的爆裂现象。上述结果说明,该微球的壳壁弹性不足,膨胀性能较差,这可能是由于TMPTA为三乙烯基的交联剂,在相同的交联剂用量条件下,TMPTA能形成更多的交联点,导致壳壁变硬所制。基于上述结果,本文又尝试了三种双乙烯基的交联剂,包括DVB、EGDMA和高分子交联剂P1。与TMPTA相比,采用双乙烯基的交联剂后,由于交联点密度下降,壳壁弹性增加,膨胀性能变好。以DVB为交联剂时,微球的发泡倍率增加至3.3倍,但须指出的是,以DVB为交联剂的微球在发泡过程中仍存在一定程度的爆裂现象。说明发泡剂的膨胀能力与微球壳壁的形变能力间匹配性仍不足,导致部分微球的壳壁在膨胀过程中破裂。
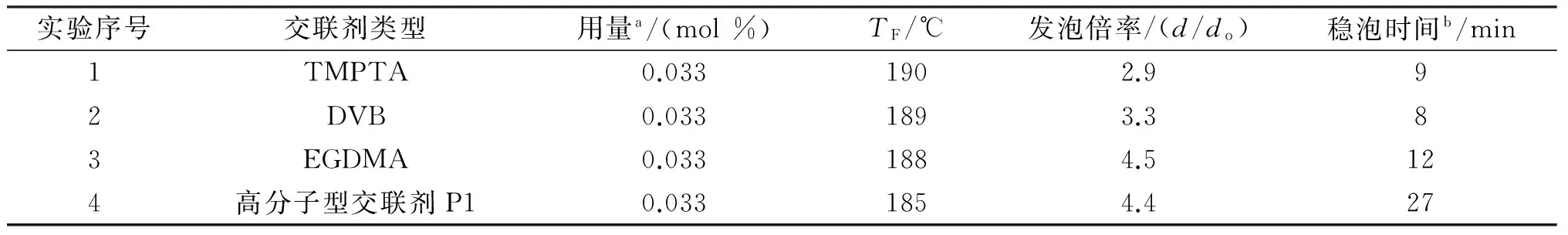
表1 交联剂类型对微球发泡性能的影响
注:a表示基于单体的总摩尔量;b表示温度为185 ℃时,发泡后微球的稳泡时间。
以EGDMA和P1为交联剂时,微球的发泡倍率显著增加到近4.5倍,且在发泡过程中未出现明显的爆裂现象。说明以EGDMA和P1为交联剂制得的微球壳壁的形变能力优异,且与发泡剂的膨胀能力匹配性较好,因此随发泡剂的膨胀,微球壳壁能均匀地产生形变,微球的发泡倍率显著增加。但需指出的是,以EGDMA为交联剂的微球虽有较好的膨胀性能,但其高温稳泡性能不佳。在185 ℃下,膨胀后的微球在12 min后就会出现明显的收缩。以P1为交联剂的微球,不仅有很好的膨胀性能,而且具有优良的高温稳泡性能,膨胀后的微球在185 ℃的烘箱中放置27 min后未出现明显的收缩。以EGDMA和P1为交联剂的微球在高温稳泡性能上的显著差异可能是因为P1为高分子交联剂,因此P1交联的分子链间距离要显著大于EGDMA交联的分子链。在相同的膨胀倍率下,以EGDMA为交联剂形成的微球壳壁聚合物链所受的内应力要高于以P1为交联剂的壳壁聚合物链,因此以P1为交联剂的微球具有更好的高温稳泡性能。综上所述,采用高分子型交联剂P1为交联剂时,所制热膨胀微球不仅具有高的膨胀性能,还具有优异的高温稳泡性能。
2.3.2 交联剂用量
表2列出了交联剂P1用量对TF、发泡倍率和稳泡时间等发泡性能的影响。由表2可知,交联剂用量对微球的发泡性能存在较大影响。随着交联剂用量的增加,微球发泡温度上升。
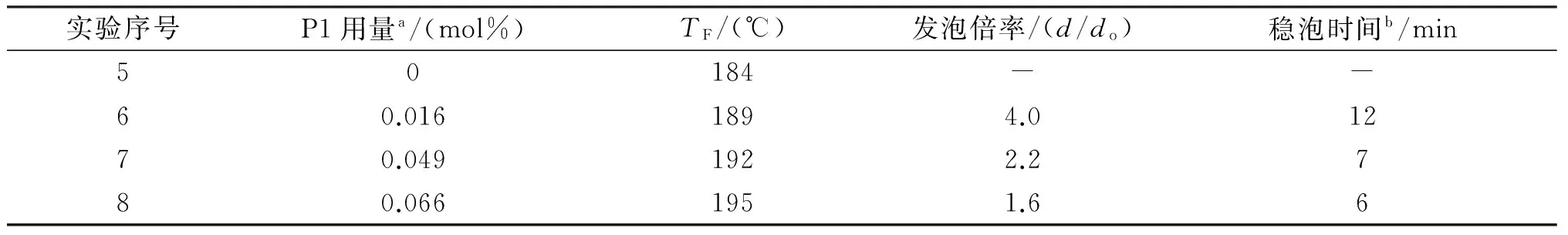
表2 高分子型交联剂P1用量对微球发泡性能的影响
注:a表示基于单体的总摩尔量;b表示温度为185 ℃时,发泡后微球的稳泡时间。
图6(a)为不同交联剂用量的样品发泡后的照片,由图可知膨胀微球的宏观发泡体积随P1用量增加呈先增大后下降的趋势。为进一步分析交联剂用量对微球发泡性能的影响,用光学显微镜观察了发泡后微球的形貌,结果见图4(b)和6(b)-(e)。由图6(b)可知,未交联的微球样品中,几乎所有的微球均能发泡,但其中也出现了较多不规则的收缩后的微球。说明该样品虽然有较好的发泡能力,但是其中较多发泡后的微球很快出现收缩,这是最终导致微球发泡后宏观体积较小的原因。由于发泡后微球稳泡性能不佳,因此无法通过统计发泡前后微球的粒径来估算该样品的发泡倍率。

图6 不同交联剂用量条件下所制微球发泡后的照片和光学显微镜照片
添加0.016 mol%的交联剂后,发泡后微球的宏观体积明显增加(图6(a)),微球的发泡倍率达到了4.0倍,说明通过引入少量交联剂后,微球仍具备较好的膨胀能力,同时稳泡性能也有了显著提升。研究发现,最佳的交联剂用量为0.033 mol%,该微球发泡后具有最大的宏观体积,发泡倍率高达4.4倍。此外,该微球的显微镜照片中未发现收缩的微球,发泡后微球的稳泡时间达到了27 min。进一步增加交联剂用量,宏观发泡体积和微球的发泡倍率均呈下降趋势。显微镜结果图6(d)和图6(e)显示高交联剂用量的微球样品中存在一定量的未发泡的微球,比如交联剂用量为0.049 mol%和0.066 mol%的微球样品中分别约有30%和60%的微球未发泡。高交联剂用量时,壳层聚合物的交联密度过大,弹性下降,使得微球发泡难度增加,导致微球的发泡倍率下降,甚至出现大量未能发泡的微球。此外,高交联剂用量的微球,由于发泡倍率小,所以在发泡温度下,微球内发泡剂膨胀所产生的内压较高,因此在微球内外压差的作用下,壳层聚合物始终受到较大的压力作用,最终导致壳壁在较短的时间内出现破裂,表现出较差的高温稳泡性能。
3 结 论
悬浮聚合法能方便地制备微米级的热膨胀微球,所制微球为非规则的球体,表面存在一定程度的凹陷。优化条件下制得的微球发泡温度为185 ℃,发泡倍率可达4.4倍,膨胀后的微球在发泡温度下稳泡时间接近30 min,具有优良的膨胀性能和高温稳泡性能。研究发现交联剂类型和用量对微球的发泡能力有较大的影响。与三乙烯基交联剂和小分子双乙烯基交联剂相比,以双乙烯基高分子型交联剂P1为交联剂时,所制微球的发泡剂膨胀能力和微球壳壁聚合物的形变能力有较好的匹配性,综合发泡性能优异。随高分子型交联剂P1用量的增加,微球的膨胀倍率和高温稳泡时间均呈先增加后下降的趋势。
在后续的工作中,将对微球壳壁的组成-发泡温度下壳壁聚合物的粘弹性-微球发泡行为间的对应关系、微球壳壁和烷烃膨胀性能的匹配性等问题展开研究,加深对微球发泡行为的理解,实现对微球发泡行为的精细调控。通过本方法制备的耐热型高温热膨胀微球可用作聚氯乙烯等塑料的添加剂,实现高分子材料的轻量化。
参考文献:
[1] Jonsson M, Nordin O, Kron A L, et al. Thermally expandable microspheres with excellent expansion characteristics at high temperature[J]. Journal of Applied Polymer Science,2010,117(1):384-392.
[2] Hu J, Zheng Z, Wang F, et al. Synthesis and characterisation of thermally expandable microcapsules by suspension polymerisation[J]. Pigment & Resin Technology,2009,38(5):280-284.
[3] Benita S. Microencapsulation: Methods and Industrial Applications[M]. 2nd Ed. Florida: CRC Press,2006.
[4] Whinnery L, Goods S, Even B. Expancel foams: Fabrication and characterization of a new reduced density cellular material for structural applications[R]. Sandia National Labs, Albuquerque, NM (US); Sandia National Labs, Livermore, CA (US),2000-08-01.
[5] Ahmad M. Flexible vinyl resiliency property enhancement with hollow thermoplastic microspheres[J]. Journal of Vinyl & Additive Technology,2001,7(3),156-161.
[6] Morehouse D S, Tetreault R J. Expansible thermoplastic polymer particles containing volatile fluid foaming agent and method of foaming the same: US3615972[P].1971-10-26.
[7] 孙伟贤,刘峰,陈宝生.可热膨胀热塑性微球:201210109302.3[P].2012-04-13.
[8] Nakayama N, Kida S. Process for preparing a thermo-expandable microspheres: US4287308 A[P].1981-09-01.
[9] Tsuda Y, Uekado K, Nakamoto H, et al. Thermal insulation
foamed material having carbon dioxide absorbents and method for manufacturing same: US 5792801[P].1998-08-11.
[10] Satake Y, Asai G. Thermally foamable microsphere and production process thereof: US 7252882 B2[P].2007-08-07.
[11] Hou Z S, Kan C Y. Preparation and properties of thermoexpandable polymeric microspheres[J]. Chinese Chemical Letters,2014,25(9):1279-1281.
[12] 何思阳,齐会民,刘峰,等.丙烯腈-丙烯酸甲酯共聚低温热膨胀微胶囊的制备与性能[J].塑料工业,2013,41(5):96-100.
[13] Safajou-Jahankhanemlou M, Abbasi F, Salami-Kalajahi M. Synthesis and characterization of thermally expandable PMMA-based microcapsules with different cross-linking density[J]. Colloid and Polymer Science,2016,294(6):1-10.
[14] Kawaguchi Y, Itamura Y, Onimura K, et al. Effects of the chemical structure on the heat resistance of thermoplastic expandable microspheres[J]. Journal of Applied Polymer Science,2005,96(4):1306-1312.
[15] Hou Z, Xia Y, Qu W, et al. Preparation and properties of thermoplastic expandable microspheres with P(VDC-AN-MMA) shell by suspension polymerization[J]. International Journal of Polymeric Materials & Polymeric Biomaterials,2015,64(8):427-431.
[16] Kawaguchi Y, Oishi T. Synthesis and properties of thermoplastic expandable microspheres: The relation between crosslinking density and expandable property[J]. Journal of Applied Polymer Science,2004,93(2):505-512.
[17] Jonsson M, Nordin O, Malmstrom E. Increased onset temperature of expansion in thermally expandable microspheres through combination of crosslinking agents[J]. Journal of Applied Polymer Science,2011,121(1):369-375.
[18] 丁婉,刘峰,缪克存,等.高温物理发泡微胶囊的制备及其对发泡性能的影响研究[J].塑料工业,2012,40(6):244-28.
[19] Jonsson M, Nordin O, Kron A L, et al. Influence of crosslinking on the characteristics of thermally expandable microspheres expanding at high temperature[J]. Journal of Applied Polymer Science,2010,118(2):1219-1229.
[20] 朱效杰,刘峰,潘肇琦,等.含甲基丙烯酸的高温发泡微球的合成及其发泡性能研究[J].塑料工业,2013,41(4):14-18.
