组学技术揭秘草食动物消化道真菌组成和功能
2018-05-11王佳堃和文凤
王佳堃,和文凤
(浙江大学奶业科学研究所,杭州310058)
真菌作为降解木质纤维素最有效的微生物类群,一直是挖掘高效纤维降解酶的核心微生物。新美鞭毛菌科(Neocallimastigaceae)是真菌界唯一营专性厌氧的类群。自1975年ORPIN[1]发现厌氧真菌是瘤胃微生物的重要组成部分以来,厌氧真菌的分类和群落结构、纤维素酶的多样性一直是微生物学和动物营养学研究的热点。随着高通量测序技术的发展,在许多生境中均检测到新美鞭毛菌科的存在,不再被误认为是仅存于草食动物消化道中;其下的属也由6个增至可分类的9个属和12个尚未分类的类群。随着真菌通用引物的使用,在反刍动物瘤胃和草食动物粪便中检测出越来越多的非严格厌氧真菌。直接饲喂厌氧真菌和酵母均能改善瘤胃内环境,提高动物生产性能,但在非外源添加情况下,非严格厌氧真菌在消化道中的作用尚未可知。为充分了解消化道中真菌的功能,本文就厌氧真菌的环境适应机制、厌氧真菌的测定方法、草食动物消化道真菌组成、非严格厌氧真菌在草食动物消化道内的作用及利用宏基因组学/宏转录组学探究消化道内真菌生理功能的瓶颈进行了综述。
1 厌氧真菌的环境适应机制
厌氧真菌可特化出氢化酶体和纤维小体结构,提高厌氧环境下自身的能量供应;也能特化出厚的细胞壁或孢子样结构增加对不良环境的适应。然而进食饲料后,厌氧真菌能迅速黏附/定殖在纤维上,但在这个过程中厌氧真菌如何防御氧的毒性尚不十分清楚。
1.1 氢化酶体的无氧代谢
线粒体是真核生物进行氧化代谢的部位,是糖类、脂肪和氨基酸最终氧化释放能量的场所,三羧酸循环与氧化磷酸化都在线粒体内完成。厌氧真菌没有线粒体,更没有线粒体内膜上的细胞色素和氧化磷酸化途径[2-3],但厌氧真菌特有的氢化酶体,是一种双层膜包裹的简单细胞器,内含氢化酶[4]。苹果酸在氢化酶作用下脱羧产生三磷酸腺苷(adenosine triphosphate,ATP)、氢气、二氧化碳和乙酸,提升了在厌氧情况下的供能效率[5-6]。鉴于氢化酶体与线粒体极其类似,被认为是线粒体的厌氧衍生物,或线粒体和氢化酶体起源于同一种兼性厌氧细菌[7]。
1.2 纤维小体提高纤维降解效率
纤维素酶是指能够水解纤维素β-1,4-D-葡萄糖苷键,使纤维素变成纤维二糖和葡萄糖的一组酶的总称。纤维素的水解需要酶预先结合到纤维素上,形成酶-底物二元复合物。好氧细菌和真菌不附着或微弱地附着纤维素,产生的酶很少形成稳定的高分子复合物,独立的胞外酶(外切β-1,4-葡聚糖酶、内切β-1,4-葡聚糖酶和β-1,4-葡萄糖苷酶)具有自己的结合域和催化域,协同降解纤维素。而厌氧微生物由于糖酵解供能效率低,所以特化出了纤维小体(cellulosome),在有限ATP的情况下,增加与其他微生物的竞争力,提高纤维物质的降解效率。纤维小体是多种纤维素酶、半纤维素酶依靠锚定-黏附机制形成的稳定高分子的多酶复合体结构,能高效降解纤维类物质[8],为糖酵解途径输送更多的葡萄糖。厌氧真菌虽然已具有氢酶体,但在进化过程中仍保留了纤维小体结构。
1.3 特化出的细胞结构和基因组对有氧损伤的防御
氧对于厌氧真菌具有毒副作用,但厌氧真菌的繁殖体可在粪便中存活1年之久[9]。NIELSEN等[10]发现,奶牛粪便中黏附在植物颗粒上的孢子有一层厚厚的壁,这些真菌孢子经过洗涤、干燥和在空气中贮存数天后,仍可再生出单中心真菌,但是瘤胃中黏附在植物颗粒上的孢子在有氧情况下却不能再生,因此他们推测,厌氧真菌在通过胃肠道时发生了细胞壁结构的变化,使其抵抗氧的毒性。TRINCI等[11]进一步指出,在有氧条件下于干燥的羊粪中可以分离到厌氧真菌,且不受贮存时间和真菌种类的影响,但在有氧且保持湿润的条件下,不论是在20℃还是39℃的羊粪中都无法分离到厌氧真菌,为此推断干燥过程可以启动真菌的防御结构。BROOKMAN等[12]也观察到一种孢子样结构,推测这种结构可使厌氧真菌在肠道厌氧环境下形成一种静止状态,有利于生长时再出芽。然而,这些均不能解释厌氧真菌在新进食纤维上快速定殖的原理。NATVIG[13]发现,在兼性的或耐氧的厌氧菌细胞内超氧化物歧化酶、过氧化氢酶和过氧化物酶的活性都低于需氧菌,而且确认壶菌纲的真菌无过氧化氢酶,但新美鞭毛菌科是否缺少上述酶系尚未可知。YOUSSEF等[3]通过对根囊鞭菌属(Orpinomyces)的C1A菌株进行单菌测序发现,在100.95 Mb的基因组中,G+C含量仅占17%,存在73.1%的非编码间隔区,4.9%的微卫星重复序列。这种特化出的基因组与防御氧损伤有何关联有待进一步研究。
2 厌氧真菌的测定方法
厌氧真菌可以通过菌体计数和酶活测定等方式定量,通过显微技术分类。然而,随着分子生物学的发展,基于核糖体18S/28SrRNA基因和转录组间隔区(internal spacer region,ITS)的分子标记技术为厌氧真菌数量和区系的研究提供了更为精准的手段,使厌氧真菌的研究逐渐深入。
2.1 显微镜观察法
显微镜很容易区分真菌的根状和球状形态,是测定真菌生长状态的最直接方法,但是区分单中心和多中心则需要采用4',6-二脒基-2-苯基吲哚(4',6-diamidino-2-phenylindole,DAPI)或双苯酰亚胺等Hoechst染料进行DNA荧光标记。但即便使用荧光标记,显微镜技术也很难区分梨囊鞭菌属(Piromyces)和新美鞭菌属(Neocallimastix)这2类单中心真菌。厌氧真菌也可依据鞭毛形态分为单鞭毛和多鞭毛,但由于根囊鞭菌属(Orpinomyces)和厌氧鞭菌属(Anaeromyces)较少释放游离孢子,所以也很难在显微镜下鉴别这2种孢子的鞭毛类型。Oontomyces和Buwchfawromyces的形态与梨囊鞭菌属极其相似,在显微镜下很难区分。Pecoramyces也一度被称作Orpinomyces sp.C1A[3],很难将其与根囊鞭菌属的真菌进行区分。此外,在荧光显微镜下植物有自发光现象,干扰了食糜碎片中真菌形态结构的识别。在表1中列出了已分离鉴定出的9个厌氧真菌属的形态特征。
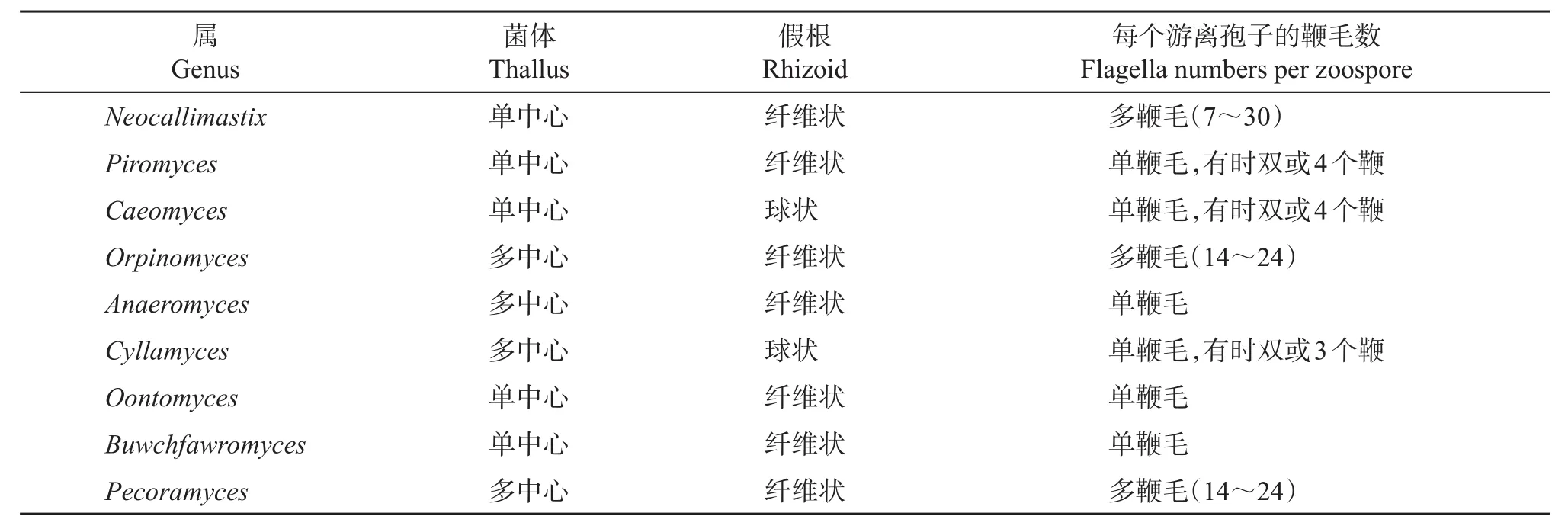
表1 9个厌氧真菌属的形态特征Table1 Morphological characteristics of ninerecognized anaerobic fungal genera
2.2 组学技术
组学技术的不断完善推动了真菌定量和区系分析的精准化。真菌核糖体内18SrDNA、内转录间隔区 1(internal transcribed spacer,ITS1)、5.8S rDNA、ITS2、28S rDNA、基因间隔区 1(intergenic spacer region,IGS1)、5SrDNA和IGS2依次串联形成多拷贝的结构(图1)[14]。基于核糖体大小亚基的结构,研究人员设计出不同的引物用于荧光定量和真菌区系分析。ITS1和28SrDNA的D1/D2区域序列联用已成为真菌区系精细分析的首选。

图1 厌氧真菌核糖体操纵子结构示意图[14]Fig.1 Schematic diagram of rrnoperon in anaerobic fungi[14]
2.2.1 荧光定量分析
18SrRNA基因由于其保守程度高达97%,所以主要用于厌氧真菌的定量。到目前为止,厌氧真菌的4对定量引物分别由以下研究人员设计提出。1)DENMAN等[15]:扩增18SrRNA基因3'端和ITS1 5'端共120 bp的区域;2)EDWARDS等[16]:扩增5.8S rRNA基因110 bp的区域;3)LI等[17]、KITTELMANN等[18]:扩增从18SrRNA基因启始部位到部分5.8S rRNA基因终止的433 bp区域;4)DOLLHOFER等[19]:扩增18SrRNA基因475 bp的长度区域。真菌DNA和生物量间的比值随真菌生长阶段不断变化,虽然LWIN等[20]利用9株厌氧真菌证实ITS1基因拷贝数与生物量和游动孢子计数之间存在显著的正相关,但上述4对引物仅能通过基因拷贝数来描述和对比不同样品或生境中厌氧真菌丰度的差异,不能真实地反映厌氧真菌的生物量。
2.2.2 多样性分析
ITS不转录成RNA,在进化过程中受到的选择压力小,容易产生变异,在绝大多数真菌的不同种类之间表现出广泛的多态性,是目前公认的真菌分子鉴定和多样性研究的分子标记物。ITS1和ITS全长序列(ITS1-5.8SrDNA-ITS2)已在变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)、限制性内切酶片段长度多态性分析(restriction fragment length polymorphism,RFLP)及宏基因组学(metagenomics)中成功应用,而且随ITS扩增引物的完善,使厌氧真菌的分类更精准,厌氧真菌的分类也增至6个属,以及12个以上尚未命名的属[21-24]。
由于ITS变异频率高,在ITS内经常有插入和缺失,所以同一种的不同菌株可能会有ITS不一致的现象。为了保证测序结果的准确,TAN等[25]推荐将18S、28S和ITS 3个RNA基因区域整合测序。DAGAR等[26]在利用ITS1/ITS4引物[27]扩增ITS(部分18S、完整的ITS1、5.8S、ITS2和部分28S序列)、利用NL1/NL4引物[28]扩增28SrDNA的D1/D2区域序列时发现,在印度骆驼前胃中分离出的2株形态与梨囊鞭菌属相似的厌氧真菌与梨囊鞭菌属的亲缘距离很远,但与厌氧鞭菌属的亲缘关系较近,将其命名为Oontomyces新属(图2)[31]。CALLAGHAN等[29]同样利用28SrDNA的D1/D2区域(DAGAR等[28]的引物NL1/NL4)及 ITS1序列(EDWARDS等[16]的引物GM1/MN106扩增产物跨ITS1和ITS2)鉴别出西威尔士的水牛、绵羊、奶牛和马的粪便中另一个形态与梨囊鞭菌属相似,但与厌氧鞭菌属亲缘关系较近的Buwchfawromyces属(图2)[31]。WANG等[30]在上述研究的基础上系统地比较了ITS1、ITS全长序列及28S rDNA D1/D2区域扩增产物的系统发育树结构,发现厌氧鞭菌属和Oontomyces的ITS1无明显的进化距离,ITS全长无法区分根囊鞭菌属的菌株,而核糖体大亚基不存在上述问题,因此推荐用核糖体大亚基进行厌氧真菌的分类鉴定。HANAFY等[31]结合镜检和28SrDNA测序,鉴别出从牛和羊胃中分离的4株厌氧真菌为Pecoramyces属。
3 草食动物消化道真菌组成
反刍动物的瘤胃、部分草食动物的前胃和盲肠的pH值接近中性,为厌氧细菌和真菌提供了良好的生存条件。但畜种和消化道类型、日粮结构均影响动物消化道真菌的组成。
3.1 畜种和消化道类型的影响
LIGGENSTOFFER等[21]利用ITS1序列,系统比较了叉角羚科、牛科、鹿科、长颈鹿科等反刍动物与前胃发酵的非反刍动物袋鼠科、伪反刍动物骆驼科和河马科以及后肠发酵动物马科、美洲鬣蜥科和犀科共29种动物粪便中厌氧真菌的组成(图3),发现厌氧真菌的OTU数在4种发酵类型的动物粪便中无显著差异,但在后肠发酵动物粪便中的厌氧真菌组成明显有别于前胃发酵类型,在黑犀牛粪便中99.9%的真菌源自于梨囊鞭菌属,斑马粪便中99.9%的真菌源自于NG1属,小型驴NG3占98.8%,索马里野驴中新美鞭菌属、盲肠鞭菌属、NG1和NG3分别占44.9%、15%、4.7%和34.7%。在前胃发酵动物粪便中新美鞭菌属、梨囊鞭菌属和盲肠鞭菌属是优势真菌属,但同一科的不同动物间也存在丰度差异。
本课题组利用BUÉE等[32]的引物扩增ITS1序列,分析了水牛、奶牛和湖羊瘤胃内容物,兔盲肠内容物,马和麋鹿粪便中真菌的多样性,并利用荧光定量聚合酶链式反应(polymerase chain reaction,PCR)测定了其菌群数量[33];同时,利用糖苷水解酶家族10和11验证了消化道内真菌的纤维分解功能[34]。研究发现,在草食动物消化道中存在大量好氧真菌,无论这些真菌是一过性还是长期定殖,它们在消化道中都行使了一定的纤维消化功能,且酵母纲所占比例高,具有潜在开发价值。
ABRÃO等[35]比较了阉公牛、奶牛和犊牛在进食同种高木质化臂形草8 h后瘤胃内真菌区系组成的差异,发现厌氧真菌在阉公牛瘤胃样品中广泛分布,而在奶牛瘤胃样品中非严格厌氧真菌量高于阉公牛,且主要是一些多中心的真菌;在阉公牛和奶牛瘤胃中最易检出的是曲霉属真菌,而在犊牛瘤胃样品中未检出厌氧真菌。在新生和哺乳动物粪便中未分离到厌氧真菌,而在成年动物粪便中分离到了厌氧真菌[36],说明厌氧真菌通过母亲的舔食或粪便随唾液或食物进入瘤胃并且存活下来。在不同畜种消化道中厌氧真菌的组成虽然不同,但是厌氧真菌在宿种间转移却有可能,如ORPIN[37]就成功地将马和鹿消化道中的厌氧真菌转移给了绵羊。

图2 草食动物消化道内厌氧真菌28Sr RNA基因序列系统发育树[31]Fig.2 Phylogenetic tree of 28SrRNA gene sequence of anaerobic fungiin the digestive tract of herbivores[31]
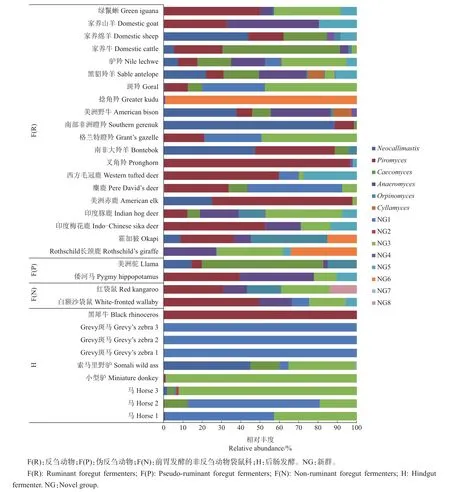
图3 草食动物消化道内厌氧真菌组成[21]Fig.3 Composition of anaerobic fungiin thedigestivetract of herbivores[21]
3.2 日粮的影响
ISHAQ等[38]利用WHITE等[39]的引物扩增ITS1序列,通过高通量分析发现,除新美鞭菌属、根囊鞭菌属、梨囊鞭菌属外,在奶牛瘤胃中存在着近50%的非严格厌氧真菌。奶牛日粮由粗料型转为高精料型诱导亚临床酸中毒后,在奶牛瘤胃中新美鞭菌属、根囊鞭菌属、梨囊鞭菌属厌氧真菌的相对丰度显著降低,而翘孢霉属(Emericella)、镰刀菌属(Fusarium)、红曲霉属(Monascus)、毕赤酵母属(Pichia)、假丝酵母(Candida)等相对丰度显著增加。
LIMA等[40]研究发现,产犊前饲喂高纤维低能量日粮,而产后饲喂低纤维高能量日粮,产前奶牛瘤胃中真菌的丰度高于产后。产犊前后,奶牛除经历日粮变化外,也经历了生理上的巨大变化,但两者影响真菌的权重尚不可知。BOOTS等[41]试验显示,6%的豆油可显著降低厌氧真菌的丰度,但日粮精粗比由50∶50增至90∶10并未显著影响厌氧真菌的丰度。KOLK等[42]利用银合欢研究了浓缩单宁对山羊瘤胃真菌数量的影响,发现饲喂浓缩单宁的前10 d,瘤胃真菌受到抑制,饲喂15 d时真菌数量增长4倍,但随后真菌数量又开始下降,在饲喂30 d时瘤胃真菌数量与未饲喂银合欢的山羊相比无显著差异。
利用荧光定量PCR技术,我们在RNA水平上对采食前后共9个时间点的瘤胃好氧和厌氧菌群进行分析,发现采食前每克瘤胃内容物中总真菌、子囊菌门(Ascomycota)真菌、新美鞭毛菌门(Neocallimastigomycota)真菌、担子菌门(Basidiomycota)真菌的拷贝数分别高达1×109、1×1×107、1×106和1×107,进食后1 h总真菌数有上升趋势,子囊菌门和担子菌门显著上升,3 h后上述真菌均有所下降,但新美鞭毛菌门真菌数量相对稳定,未随采食时间的变化出现显著波动[43]。
4 非严格厌氧真菌在草食动物消化道内的作用
利用真菌通用引物,在草食动物消化道内越来越多的非严格厌氧真菌被发现和报道,其中包括子囊菌门、担子菌门和接合菌门(Zygomycota),而且在瘤胃中子囊菌门相对丰度可高达22.6%[44]。直接饲喂瘤胃厌氧真菌可以提高反刍动物的采食量、乳质量和乳产量等[45-46]。酵母(Saccharomycetes)作为子囊菌门中的重要成员,饲喂活酵母可促进瘤胃微生物数量和活力,改善产肉和产奶性能也近乎是公认的结果[47],尤其是在高精料日粮下,活酵母可以加速瘤胃乳酸代谢,稳定瘤胃pH值[48]。CHAUCHEYRASDURAND等[49]的研究证实,饲喂活酵母可改善瘤胃的发酵特性,增强瘤胃微生物对纤维饲料的定殖,进而提高微生物对纤维饲料的降解能力。PINLOCHE等[50]和ALZAHAL等[51]也证实活酵母可稳定瘤胃的pH值,降低氧化还原电位和乳酸浓度,提高纤维分解菌(纤维杆菌属Fibrobacter和瘤胃球菌属Ruminococcus)、乳酸利用菌(巨型球菌属Megasphaera和月形单胞菌属Selenomonas)、双歧杆菌(Bifidobacterium)的相对丰度,降低普雷沃菌(Prevotella)、互养球菌属(Syntrophococcus)、光岗菌属(Mitsuokella)的相对丰度。而且,添加酵母能增加瘤胃真菌的丰度。在高纤维日粮下,干酵母可增加Lewia、新美鞭菌属和茎点霉属(Phoma)的丰度,降低链格孢属(Alternaria)、假丝酵母属(Candida)、根囊鞭菌属和梨囊鞭菌属的丰度。
5 宏基因组学/宏转录组学探究消化道内真菌生理功能的瓶颈
宏基因组学/宏转录组学在研究瘤胃微生物代谢和开发瘤胃微生物资源上展示出强劲的优势。但由于现有的数据库(NCBIBLAST、InterProScan、OrthoMcL、KEGG、TrEMBL和SwissProt)缺少真菌的信息,所以导致真菌转录组测序后能够得以注释的基因往往不到50%[52]。在消化道微生物区系中存在细菌、真菌、原虫,且细菌占比高于真菌,增加了在消化道微生物区系中获取足量的真菌DNA/RNA用于宏基因组/宏转录组分析的难度。FERRER等[53]对奶牛瘤胃宏基因组文库进行了功能筛选,发现在可识别的编码序列中厌氧真菌仅占了5%;而DAI等[54]发现,瘤胃微生物在宏转录组测序数据中厌氧真菌的序列数仅占总序列数的1%。
6 小结
分子生物学技术的发展使消化道中真菌的定量和区系分析越来越精准,而且越来越多的真菌酶基因被扩增和异源表达,开发出新酶用于工业生产。但消化道中真菌的生理、厌氧真菌与非严格厌氧真菌的互作等尚未清楚,宏基因组学/宏转录组学技术必然是有利的推手。因此,突破宏基因组学/宏转录组学探究消化道内真菌生理功能的瓶颈是今后真菌研究的重中之重。
参考文献(References):
[1] ORPIN C G.Studies on the rumen flagellate Neocallimastix frontalis.Microbiology,1975,91:249-262.
[2]YARLETT N,ORPIN C G,MUNN E A,et al.Hydrogenosomes in the rumen fungus Neocallimastix patriciarum.Biochemical Journal,1986,236(3):729-739.
[3]YOUSSEF N H,COUGER M B,STRUCHTEMEYER C G,et al.The genome of the anaerobic fungus Orpinomyces sp.strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Applied and Environmental Microbiology,2013,79(15):4620-4634.
[4]HACKSTEIN JH P,AKHMANOVAA,VONCKEN F,et al.Hydrogenosomes:Convergent adaptations of mitochondria to anaerobic environments.Zoology,2001,104(3/4):290-302.
[5] MULLER M.The hydrogenosome.Microbiology,1993,139(12):2879-2889.
[6]MÜLLER M,MENTEL M,VAN HELLEMOND JJ,et al.Biochemistry and evolution of anaerobic energy metabolism in eukaryotes.Microbiology and Molecular Biology Reviews,2012,76(2):444-495.
[7]VAN HOEK A H A M,VAN ALEN T A,SPRAKEL V SI,et al.Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates.Molecular Biology and Evolution,2000,17(2):251-258.
[8]XU Q,BAYER E A,GOLDMAN M,et al.Architecture of the Bacteroides cellulosolvens cellulosome:Description of a cell surface-anchoring scaffoldin and a family 48 cellulase.Journal of Bacteriology,2004,186(4):968-977.
[9]MCGRANAGHAN H,DAVIESJC,GRIFFITH G W,et al.The survival of anaerobic fungi in cattle faeces.FEMS Microbiology Ecology,1999,29(3):293-300.
[10]NIELSEN B B,ZHU W Y,TRINCI A P J,et al.Demonstration of zoosporangia of anaerobic fungi on plant residues recovered from faeces of cattle.Mycological Research,1995,99(4):471-474.
[11]TRINCI A P J,LOWE S E,MILNE A,et al.Growth and survival of rumen fungi.Biosystems,1988,21(3/4):357-363.
[12]BROOKMAN J L,MENNIM G,TRINCI A P J,et al.Identification and characterization of anaerobic gut fungi using molecular methodologies based on ribosomal ITS1 and 18SrRNA.Microbiology,2000,146(2):393-403.
[13]NATVIG D O.Comparative biochemistry of oxygen toxicity in lactic acid-forming aquatic fungi.Archives of Microbiology,1982,132(2):107-114.
[14]EDWARDSJE,FORSTER R J,CALLAGHAN T M,et al.PCR and omics based techniques to study the diversity,ecology and biology of anaerobic fungi:Insights,challenges and opportunities.Frontiers in Microbiology,2017,8:1657.
[15]DENMAN SE,MCSWEENEY C S.Development of a realtime PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen.FEMS Microbiology Ecology,2006,58(3):572-582.
[16]EDWARDSJE,KINGSTON-SMITH A H,JIMENEZ H R,et al.Dynamics of initial colonization of nonconserved perennial ryegrass by anaerobic fungi in the bovine rumen.FEMSMicrobiology Ecology,2008,66(3):537-545.
[17]LI J,HEATH I B.The phylogenetic relationships of the anaerobic chytridiomycetous gut fungi(Neocallimasticaceae)and the Chytridiomycota.Ⅰ.Cladistic analysis of rRNA sequences.Canadian Journal of Botany,1992,70(9):1738-1746.
[18]KITTELMANN S,NAYLOR G E,KOOLAARD J P,et al.A proposed taxonomy of anaerobic fungi(Class Neocallimastigomycetes)suitable for large-scale sequence-based community structure analysis.PLoSOne,2012,7(5):e36866.
[19]DOLLHOFER V,CALLAGHAN T M,DORN-IN S,et al.Development of three specific PCR-based tools to determine quantity,cellulolytic transcriptional activity and phylogeny of anaerobic fungi.Journal of Microbiological Methods,2016,127:28-40.
[20]LWIN K O,HAYAKAWA M,BAN-TOKUDA T,et al.Realtime PCR assays for monitoring anaerobic fungal biomass and population size in the rumen.Current Microbiology,2011,62(4):1147-1151.
[21]LIGGENSTOFFER A S,YOUSSEF N H,COUGER M B,et al.Phylogenetic diversity and community structure of anaerobic gut fungi(phylum Neocallimastigomycota)in ruminant and non-ruminant herbivores.The ISME Journal,2010,4(10):1225-1235.
[22]NICHOLSON M J,MCSWEENEY C S,MACKIE R I,et al.Diversity of anaerobic gut fungal populations analysed using ribosomal ITS1 sequences in faeces of wild and domesticated herbivores.Anaerobe,2010,16(2):66-73.
[23]GRUNINGER R J,PUNIYA A K,CALLAGHAN T M,et al.Anaerobic fungi(phylum Neocallimastigomycota):Advances in understanding their taxonomy,life cycle,ecology, role and biotechnological potential. FEMS Microbiology Ecology,2014,90(1):1-17.
[24]KOETSCHAN C,KITTELMANN S,LU J L,et al.Internal transcribed spacer 1 secondary structure analysis reveals a common core throughout the anaerobic fungi(Neocallimastigomycota).PLoSOne,2014,9(3):e91928.
[25]TAN H M,CAO L X.Fungal diversity in sheep(Ovis aries)and cattle(Bos taurus)feces assessed by comparison of 18S, 28S and ITS ribosomal regions.Annals of Microbiology,2015,64(3):1423-1427.
[26]DAGAR S S,KUMAR S,GRIFFITH G W,et al.A new anaerobic fungus(Oontomyces anksri gen.nov.,sp.nov.)from the digestive tract of the Indian camel(Camelus dromedarius).Fungal Biology,2015,119(8):731-737.
[27]FLIEGEROVA K,MRAZEK J,VOIGT K.Differentiation of anaerobic polycentric fungi by rDNA PCR-RFLP.Folia Microbiologica,2006,51(4):273-277.
[28]DAGAR SS,KUMAR S,MUDGIL P,et al.D1/D2 domain of large-subunit ribosomal DNA for differentiation of Orpinomyces spp.Applied and Environmental Microbiology,2011,77(18):6722-6725.
[29]CALLAGHAN T M,PODMIRSEG SM,HOHLWECK D,et al.Buwchfawromyces eastonii gen.nov.,sp.nov.:A new anaerobic fungus(Neocallimastigomycota)isolated from buffalo faeces.Mycokeys,2015,9:11-28.
[30]WANG X W,LIU X Z,GROENEWALD JZ.Phylogeny of anaerobic fungi(phylum Neocallimastigomycota),with contributions from yak in China.Antonie Van Leeuwenhoek,2017,110(1):87-103.
[31]HANAFY RA,ELSHAHED M S,LIGGENSTOFFERA S,et al.Pecoramyces ruminantium,gen.nov.,sp.nov.,an anaerobic gut fungus from the feces of cattle and sheep.Mycologia,2017,109(2):231-243.
[32]BUÉEM,REICH M,MURANT C,et al.454 pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity.New Phytologist,2009,184(2):449-456.
[33]孙中远.反刍动物与单胃草食动物消化道氢代谢微生物差异研究.杭州:浙江大学,2013.SUN Z Y.Study on hydrogen metabolism microbiota in the gastrointestinal tract of ruminants and monogastric herbivorous animals.Hangzhou:Zhejiang University,2013.(in Chinese with English abstract)
[34]罗阳.基于宏基因组技术发掘动物消化道微生物新型木聚糖酶基因.杭州:浙江大学,2015.LUOY.Metagenomic approach to apply novel xylanase genes in animals’gastrointestinal microbes.Hangzhou:Zhejiang University,2015.(in Chinesewith English abstract)
[35]ABRÃO F O,DUARTE E R,FREITAS C E S,et al.Characterization of fungi from ruminal fluid of beef cattle with different ages and raised in tropical lignified pastures.Current Microbiology,2014,69(5):649-659.
[36]FONTY G,GOUTEP,JOUANY JP,et al.Establishment of the microflora and anaerobic fungi in the rumen of lambs.Microbiology,1987,133:1835-1843.
[37]OPRIN C G.Ecology of rumen anaerobic fungi in relation to the nutrition of the host animal//The Roles of Protozoa and Fungi in Ruminant Digestion.Armidale,Australia:Penambul Books,1989:29-38.
[38]ISHAQ SL,JOHNSON SP,MILLER Z J,et al.Impact of cropping systems,soil inoculum,and plant species identity on soil bacterial community structure.Microbial Ecology,2016,73(2):417-434.
[39]WHITE T J,BRUNS T,LEE S,et al.Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics//INNISM A,GELFAND D H,SNINSKY JJ.PCR Protocols:A Guide to Methods and Applications.San Diego,USA:Academic Press,Inc.,1990:315-322.
[40]LIMA F S,OIKONOMOU G,LIMA S F,et al.Prepartum and postpartum rumen fluid microbiomes:Characterization and correlation with production traits in dairy cows.Applied and Environmental Microbiology,2015,81(4):1327-1337.
[41]BOOTS B,LILLIS L,CLIPSON N,et al.Responses of anaerobic rumen fungal diversity (phylum Neocallimastigomycota)to changes in bovine diet.Journal of Applied Microbiology,2013,114(3):626-635.
[42]KOLK CM,SIEOCC,TAN HY,et al.Anaerobic cellulolytic rumen fungal populations in goats fed with and without Leucaena leucocephala hybrid,as determined by real-time PCR.Journal of Microbiology,2013,51(5):700-703.
[43]和文凤.饲料源真菌对瘤胃真菌组成的影响.杭州:浙江大学,2018.HE W F.Effects of feed source fungi on rumen fungus composition.Hangzhou:Zhejiang University,2018.(in Chinese with English abstract)
[44]ZHANG J,SHI H T,WANG Y J,et al.Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers.Frontiers in Microbiology,2017,8:2206.
[45]DEY A,SEHGAL JP,PUNIYA A K,et al.Influence of an anaerobic fungal culture(Orpinomyces sp.)administration on growth rate,ruminal fermentation and nutrient digestion in calves.Asian-Australasian Journal of Animal Sciences,2004,17(6):820-824.
[46]THAREJA A,PUNIYA A K,GOEL G,et al.In vitro degradation of wheat straw by anaerobic fungi from small ruminants.Archivesof Animal Nutrition,2006,60(5):412-417.
[47]WALLACE R J.Rumen microbiology,biotechnology and ruminant nutrition:The application of research findings to a complex microbial ecosystem.FEMSMicrobiology Letters,1992,100(1/2/3):529-534.
[48]WILLIAMSPE,TAITCAG,INNESGM,et al.Effectsof the inclusion of yeast culture(Saccharomyces cerevisiae plus growth medium)in the diet of dairy cows on milk yield and forage degradation and fermentation patterns in the rumen of steers.Journal of Animal Science,1991,69(7):3016-3026.
[49]CHAUCHEYRAS-DURAND F,AMEILBONNEA,BICHAT A,et al.Live yeasts enhance fibre degradation in the cow rumen through an increase in plant substrate colonisation by fibrolytic bacteriaand fungi.Journal of Applied Microbiology,2016,120(3):560-570.
[50]PINLOCHE E,MCEWAN N,MARDEN J P,et al.The effects of a probiotic yeast on the bacterial diversity and population structure in the rumen of cattle.PLoSOne,2013,8(7):e67824.
[51]ALZAHAL O,DIONISSOPOULOSL,LAARMANA H,et al.Active dry Saccharomyces cerevisiae can alleviate the effect of subacute ruminal acidosis in lactating dairy cows.Journal of Dairy Science,2014,97(12):7751-7763.
[52]SOLOMONK V,HAITJEMA CH,HENSKEJK,et al.Earlybranching gut fungi possess a large,comprehensive array of biomass-degrading enzymes.Science,2016,351(6278):1192-1195.
[53]FERRER M,GOLYSHINA O V,CHERNIKOVA T N,et al.Novel hydrolase diversity retrieved from ametagenome library of bovinerumenmicroflora.Environmental Microbiology,2005,7(12):1996-2010.
[54]DAIX,TIANY,LIJT,et al.Metatranscriptomic analyses of plant cell wall polysaccharidedegradation by microorganisms in the cow rumen.Applied and Environmental Microbiology,2015,81(4):1375-1386.
