c-JNK信号通路对糖尿病大鼠心肌钾通道重构的氧化还原调控机制
2018-05-10李学永孙毅郑明奇石克威曾伟卜雪芹孙贺建胡占军刘刚
李学永,孙毅,郑明奇,石克威,曾伟,卜雪芹,孙贺建,胡占军,刘刚
糖尿病(DM)可引起心肌细胞钾通道(Kv)重构,特别对瞬时外向钾电流(Ito)产生影响[1],这会导致心肌动作电位时程延长和不应期改变[2-3],同时也会致使Ca2+调节障碍及收缩减弱,从而使心肌收缩性和电兴奋性异常,易导致心律失常和心力衰竭等心血管并发症[4]。最近有研究指出,DM诱发的电压门控性Kv重构是由心肌细胞氧化应激和氧化还原状态转换所介导的,存在于胞质中由硫氧还蛋白(Trx)、硫氧还蛋白还原酶(TrxR)和还原型辅酶组成的Trx系统,可使Kv处于稳定的还原状态,在Kv调节过程中有重要作用。目前对DM心肌氧化应激通过Trx系统介导心肌细胞Kv重构的研究较少,引起Kv通道重构及Ito密度下降的细胞内信号机制尚不清楚。在多种细胞中,氧化应激均可作为一种代偿性反应增加多种酶系统的活性,其中包括JNK[5-6]。JNK信号传导通路是MAPK信号通路家族成员之一。JNK是广泛存在于细胞内的一类丝氨酸/苏氨酸蛋白激酶,是细胞外界信号从细胞表面转导到细胞核内部的重要传递者,在众多的疾病进展过程中起着重要的作用[7]。SP600125是一种常用的JNK1信号通路高选择性抑制剂,可以通过竞争性结合ATP位点抑制JNK1活性。本研究从分子生物学和电生理方面观察DM对左室心肌细胞氧化还原水平和复极Ito密度的影响,探索细胞凋亡信号调节酶c-JNK在Kv通道重构中的作用,以期为DM心血管并发症的发病机制寻找一个新的突破点,从而为预防DM患者心脏功能下降和心律失常提供更有效地临床指导。
1 材料与方法
1.1 主要试剂及配制 Tyrode液、无钙Tyrode液、DMEM离体心脏灌流液,胶原酶Ⅰ、Ⅳ,蛋白酶E(Sigma公司,美国);小牛血清白蛋白(BSA,Roche公司,瑞士)。测定Ito离子流的电极内液(mmol/L):天门冬氨酸钾(K-aspartate)130,MgCl22.0,依他酸(EGTA)11,Na-HEPES 10,CaCl20.5,Na2ATP 2.0,Na-GTP 0.1,用KOH调节pH至7.1~7.2。测定Ito离子流的灌流液(mmol/L):NaCl 132,KCl 4.0,CaCl22.0,MgSO41.2,HEPES 20,Glucose1.1,用NaOH调节pH至7.35。TE缓冲液(mmol/L):Tris-HCl 10,EDTA 1,pH 7.4。
1.2 实验对象及分组 健康SD大鼠由河北医科大学实验动物中心提供,雄性,6~8周龄,体重180~200g。将45只SD大鼠随机分为DM组(n=25)和对照组(n=20)。DM组大鼠给予高糖高脂饮食饲养,单次腹腔注射链尿佐菌素(STZ)柠檬酸缓冲液65mg/kg,3d后采集尾静脉血测量血糖,血糖浓度在16.7~25.0mmol/L认为建立DM模型成功,STZ诱导成模共24只。对照组大鼠普通饮食饲养,单次腹腔注射等体积的柠檬酸缓冲液。4周时DM组2只死亡,最后进入实验DM组22只,对照组20只,作为相关实验DM组及对照组的取材动物。
1.3 左室心肌细胞分离 采用胶原酶分解法获得单个分离的左室心肌细胞。大鼠称重并测血糖后用3%戊巴比妥钠腹腔注射进行麻醉,开胸迅速剪下心脏并置于4℃肝素化的正常Tyrode液中清洗修剪,沿其主动脉走行方向插入灌注导管,用Langendorff仪器沿主动脉逆向灌流心脏(37℃,90cmH2O),使用D M E M灌流液,灌流液通以混合气体(5%CO2、95%O2)。清洗灌流2~3min至心脏停止跳动,冠状动脉变白,流出的液体变清亮。采用灌流酶液消化心脏15~20min,当流下的酶液滴数在100滴/min以上时,肉眼可见大鼠心脏胀大蓬松呈半透明状,出现拉丝或者即将拉丝,此时停止消化。心脏消化完成后用50ml低钙Tyrode液(含0.02mmol/L的CaCl2)冲洗残留酶液,约3min可终止消化。剪下左室和室间隔部的心室组织并将其置于烧杯中,烧杯中预先倒入适量含有0.1%BSA的无钙Tyrode液,用眼科剪细细剪碎心肌组织,然后用粗胶头吸管轻轻吹打数次,36~37℃静置温孵5~10min,弃上清液后得到心肌细胞。将分离的左室心肌细胞放在细胞贮存液中,置于37℃孵育箱备用。
1.4 JNK活性测定 通过测定DM大鼠左室及室间隔心肌组织中的JNK磷酸化作用强度,来反映JNK激酶的活性度。采用非放射性JNK激酶分析元件(Cell Signaling Technology公司)进行测定。用裂解缓冲液将灌流消化后的左心室和室间隔部组织样本分别制备成匀浆,4℃下15 000×g高速离心30min,提取上清液,留取4种测试样本(对照左室组,n=4;DM左室组,n=4;对照室间隔组,n=4;DM室间隔组,n=4),采用二喹啉甲酸(BCA)蛋白质测定法对蛋白质浓度进行测定。取0.4mg蛋白质用20μl固定化c-Jun融合蛋白颗粒悬浮液在4℃下孵化过夜;将免疫沉淀物用裂解缓冲液洗2次,再用酶缓冲液洗2次;再加入0.2mmol/L ATP,30℃下反应30min;加入4×Laemmli样本缓冲液终止酶反应。取10μl上清液进行SDS-PAGE电泳,并用磷酸化c-Jun(Ser63)抗体检测磷酸化c-Jun条带,应用UVP生物成像系统进行分析,并使用多测量软件(FujiF-ilm,Valhalla,NY)对结果进行量化,对比两组左室及室间隔心肌组织中的JNK磷酸化强度。
1.5 全细胞膜片钳记录Ito电流 应用美国Axon公司的200B膜片钳放大器。将玻璃微管(Sutter Instruments,Novato,CA)拉至内径为1~2μmol/L的微电极,微电极(电阻2~5MΩ)内充满电极内液,小心连接于电极支持装置上,与膜片钳放大器相连接。将数滴心室肌细胞悬液滴于带有显微镜的工作台上的灌流槽中,沉淀5~10min,待细胞沉底附壁后,用95%O2和5%CO2混合气充分饱和过的灌流液(Ito电极外液)以1~2ml/min的速度恒速灌流,冲去死亡细胞碎片。在显微镜下选用形态良好的心肌细胞进行电生理记录。将内充电极内液的玻璃微电极与细胞封接、破膜,形成全细胞记录模式(环境温度22~25℃),放置5min以上以确保微电极内的电极内液与细胞质混合均匀。标准的细胞内外液将被用于所有的试验,在记录的细胞外液添加5μmol/L硝苯地平阻断Ca2+通道。使用电脑程序调控诱发电位并捕捉电流信号(经2kHz过滤处理)。样本信号取自4kHz经12位分辨率数模转换器转换,然后储存在电脑中。刺激方案:①通过计算机发放电压2mV、脉宽10ms的连续刺激脉冲补偿电容电流;②保持电位–80mV、100ms预脉冲到–60mV失活快钠通道;③Ito刺激脉宽为500ms,刺激电压范围为–40mV~+60mV,阶跃10mV,频率0.2Hz;④Ito幅值取+60mV时电流峰值。每次测试脉冲所产生的Ito电流振幅(外向电流峰电位与去极化电流末期的稳定电位之差)均加以记录。电流峰值除以被记录细胞电容大小为该心肌细胞的电流密度,单位为pA/pF。
在显微镜下选用形态良好的心肌细胞进行电生理记录,在标准的全细胞膜片方案电压钳模式下对对照组及DM组大鼠的心肌细胞进行刺激,应用软件(Pulse/PulseFit,V.8.11,HEKA Elektronik)采集Ito数据,记录电流图,计算Ito密度(单位:pA/pF),对照组成功获得16个心肌细胞Ito电流图(对照组,n=16),DM组成功获得17个心肌细胞Ito电流图(DM组,n=17),观察DM对大鼠心肌细胞Ito密度的影响。
1.6 观察JNK对DM大鼠心肌电生理的影响 在离体心肌中使用标准的全细胞膜片方案电压钳检验JNK激酶抑制剂对Ito的影响。DM大鼠心肌细胞经JNK抑制剂SP600125(10μmol/L)处理4~5h后,测量Ito密度,成功获得18个此类DM心肌细胞Ito电流图(DM+SP600125组,n=18);将DM大鼠心肌细胞经膜渗透性蛋白抑制剂JNKI-1(10μmol/L)处理4~5h后,成功获得10个此类DM心肌细胞的Ito电流图(DM+JNKI-1组,n=10);用无活性的膜渗透性蛋白抑制剂JNKI-Neg(10μmol/L)处理DM大鼠心肌细胞4~5h后,成功获得7个此类DM心肌细胞的Ito电流图(DM+JNKI-Neg组,n=7)。将得到的Ito密度值进行比较。
1.7 观察TrxR抑制剂对DM大鼠心肌的Ito电流强度的影响 为了检验Trx系统是否参与了JNK激酶对心肌Kv通道重构的调控,采用TrxR抑制剂金诺芬(AF,10nmol/L)对DM大鼠心肌细胞进行4~5h处理再测量Ito密度,成功获得13个此类DM心肌细胞的Ito电流图(DM+AF组,n=13),与未经AF处理的对照组(n=16)Ito密度进行比较;应用AF对经SP600125处理过的DM大鼠心肌细胞进行4~5h处理后再测量Ito密度,观察处理前后Ito密度的变化,成功获得15个此类DM心肌细胞的Ito电流图(DM+SP600125+AF组,n=15),观察TrxR对JNK的影响。对照组大鼠心肌细胞分别应用AF和SP600125进行4~5h处理后测量Ito密度,观察处理前后Ito密度的变化,分别成功获得18个和12个对照组心肌细胞的Ito电流图(对照+SP600125组,n=18:对照+AF组,n=12),观察二者对正常心肌的影响。
1.8 Western blotting法检测Kv4.2含量 检测JNK抑制剂SP600125对Kv4.2(Ito电流的主要亚基)表达的影响。将灌流消化后的左室心肌组织样本放入低温、已加入蛋白酶抑制混合物的TE缓冲液中并摇匀,置于离心机离心(1000×g,10min)以除去细胞核和碎片,然后再次将颗粒悬浮于TE缓冲液中,重新摇匀并离心。将两次低速离心的上清液融合并高速离心(40 000×g,15min),将沉淀的颗粒重新悬浮于TE缓冲溶液(已加入600mmol/L碘化钾以去除心肌蛋白),之后重新高速离心(40 000×g,10min)。将沉淀颗粒用TE溶液洗2次,溶解于含2%Triton X-100的TE缓冲液中并冷冻1h。之后将不溶物利用高速离心(15 000×g,15min)去除。留取对照组和DM组左室心肌组织样本,各分为8份,然后将两种样本各取出4份,分别经SP600125(10μmol/L)处理4~5h,则得4种样本各4份(对照组、DM组、对照+SP600125组、DM+SP600125组),将所有检测样本蛋白质等量行SDS-PAGE,然后转膜至聚偏氟乙烯膜。使用含有5%脱脂奶粉的商业缓冲液(Pierce,Rockford,IL)室温封闭1h。使用抗Kv4.2抗体(Alamone Labs,Tel Aviv,Israel)对Kv4.2的含量进行检测。采用高级免疫印迹观测设备(GE Healthcare,Piscataway,NJ)对结果进行观测,使用UVP生物成像系统进行分析,并使用多测量软件(FujiFilm,Valhalla,NY)对结果进行量化,所得数据进行比较。
1.9 统计学处理 采用SPSS 18.0软件进行统计分析。计量数据均以表示,两组间比较采用t检验,多组间比较采用方差分析,进一步两两比较采用LSD-t检验。P<0.05为差异有统计学意义。
2 结 果
2.1 DM大鼠心肌细胞JNK活性的检测 与对照组相比,DM组磷酸化JNK(p-JNK)含量明显升高(左室:2.2±0.4倍;室间隔:2.0±0.2倍;图1)。
2.2 DM对大鼠心肌细胞Ito密度的影响 全细胞膜片钳记录结果显示,0~60mV时,DM组大鼠心肌细胞Ito密度均明显低于对照组(P<0.05,图2)。

图1 DM大鼠心脏JNK活性(Western blotting)Fig.1 JNK activity of diabetic rat heart (Western blotting)
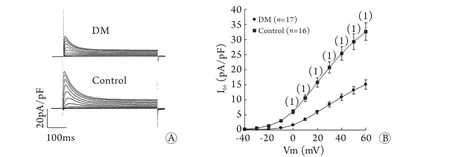
图2 糖尿病对大鼠心肌细胞Ito密度的影响Fig.2 Effect of diabetic on Ito density of cardiomyocytes in rats
2.3 JNK对DM大鼠心肌电生理的影响 结果显示,JNK抑制剂SP600125明显增加了Ito电流密度(图3A)。DM+SP600125组的Ito电流密度(60mV时)为32.3±3.7pA/pF,与DM组(15.3±2.1pA/pF)比较明显增强(P<0.05,图3B)。DM大鼠心肌经膜渗透性蛋白抑制剂JNKI-1(10μmol/L)处理后,Ito密度也明显增加(DM+JNKI-1组:28.6±3.1pA/pF),而经无活性的膜渗透性蛋白抑制剂(JNKI-Neg,10μmol/L)处理后Ito密度无明显改变(DM+JNKI-Neg组:15.8±2.4pA/pF)。

图3 JNK抑制剂对Ito密度的影响Fig.3 Upregulation of Ito density by JNK inhibitors
2.4 TrxR抑制剂对DM大鼠心肌Ito密度的影响为了检验硫氧还蛋白系统是否参与了JNK激酶对心肌Kv通道重构的调控,本研究检测了TrxR抑制剂AF对DM大鼠心肌Ito密度的影响,结果显示,AF明显抑制了SP600125对DM大鼠心肌Ito的增强作用(DM+SP600125+AF组:15.7±3.3pA/pF;DM+SP600125组:32.3±3.7pA/pF;P<0.05),而AF对对照组Ito无明显影响(对照组:30.2±3.3pA/pF;对照+AF组:31.9±3.5pA/pF)。此外,DM组中的SP600125处理组最大Ito密度(DM+SP600125组:32.3±3.7pA/pF)与对照组(30.2±3.3pA/pF)比较差异无统计学意义。且对照组经SP600125或SP600125+AF处理后的最大Ito密度(对照+SP600125组:31.6±3.4pA/pF,对照+SP600125+AF组:31.2±3.7pA/pF)与未经处理的对照组差异无统计学意义(表1)。
2.5 JNK抑制剂对Kv4.2表达的影响 Western blotting检测结果显示,与对照组相比,未经处理的DM大鼠心肌(DM组)Kv4.2蛋白丰度明显降低(P<0.05)。但经SP600125处理后,尽管未完全恢复到对照组水平,DM+SP600125组心肌的Kv4.2蛋白表达量与DM组比较明显增加(P<0.05)。对照组经SP600125处理后,Kv4.2蛋白表达无明显变化,与先前在DM心肌所观察到的Ito电流改变一致(图4)。
表1 AF对DM大鼠心肌Ito密度的影响(±s,pA/pF)Tab.1 Effect of AF on Ito density of cardiomyocytes in DM rats (±s, pA/pF)

表1 AF对DM大鼠心肌Ito密度的影响(±s,pA/pF)Tab.1 Effect of AF on Ito density of cardiomyocytes in DM rats (±s, pA/pF)
AF. Auranofin; (1)P<0.05 compared with DM group; (2)P<0.05 compared with DM+SP600125 group
Group –40mV –20mV 0mV 20mV 40mV 60mV Control (n=16) 0.2±0.1 1.2±0.3 6.4±0.9 16.0±1.9 24.8±2.8 30.2±3.3 Control+SP600125 (n=18) 0.2±0.1 1.3±0.2 6.5±1.1 16.4±1.8 25.6±2.9 31.6±3.4 Control+AF (n=12) 0.2±0.1 1.3±0.3 6.2±0.7 16.2±2.0 25.3±3.1 31.9±3.5 Control+SP600125+AF (n=12) 0.2±0.1 1.2±0.3 6.6±0.9 14.9±1.6 24.5±3.0 31.2±3.7 DM (n=17) 0.2±0.1 0.6±0.2 3.5±0.6 8.2±1.2 12.1±1.9 15.3±2.1 DM+SP600125 (n=18) 0.2±0.1 1.2±0.3(1) 6.5±0.8(1) 15.3±1.4(1) 24.3±3.2(1) 32.3±3.7(1)DM+AF (n=13) 0.2±0.1 0.7±0.2 3.5±0.5 9.1±0.9 12.5±1.7 15.1±2.3 DM+SP600125+AF (n=15) 0.2±0.1 0.6±0.2(2) 3.7±0.6(2) 9.2±1.1(2) 13.1±1.6(2) 15.7±3.3(2)

图4 SP600125对DM大鼠心肌Kv4.2蛋白表达的影响Fig.4 Upregulation of DM myocardial Kv4.2 protein expression by SP600125
3 讨 论
DM可引起心肌电重构,表现为ECG异常和多种Kv变化[1]。在啮齿类动物心肌中,由疾病所致Kv通道的减少会导致动作电位时程(APD)异常延长和不应期改变[8]。APD由细胞内、外向各种离子通道电流相互之间的精确平衡所控制,心肌细胞膜内向离子电流的增加和(或)外向电流的减少都会延长APD及QTc间期。细胞膜上的Ito是心肌细胞动作电位(AP)复极的主要电流,Ito的降低不仅导致APD延长,还会致使Ca2+调节障碍及收缩减弱,影响心肌的APD及兴奋、收缩和传导功能,ECG可出现QTc间期、QRS时限、ST段水平、T波形态等的改变,继而影响心脏的功能,出现左心室缩短分数(LVFS)和左心室射血分数(LVEF)下降[3]。而APD、QRS波时限和QTc间期延长均易导致心律失常的发生[9]。然而,引发Kv通道表达及Ito密度下降的细胞机制尚未完全明了。
研究发现,氧化应激在DM及其心血管并发症中发挥着重要作用,尤其是在心肌离子通道Kv4的重构中起着关键作用,DM诱导的心律失常和心室功能障碍常与氧化应激损害密切相关[3,9-10]。在高糖环境下,人体内环境中的各种氧化物质浓度升高,抗氧化物质的抑制因子增多,常出现Trx-1分子表达不足,导致机体抗氧化能力降低,从而引起细胞内氧化应激水平明显升高[9],其主要机制是发生在含有半胱氨酸的巯基蛋白质上的可逆性氧化修饰[11],此类蛋白中包含大量维持细胞稳态的调控信息,可影响细胞膜离子通道的功能。Trx系统和Grx系统作为重要的内源性巯基转移酶,是心肌细胞内氧化还原调控的主要组成部分,通过催化蛋白质中巯基二硫键的转换反应,保持细胞内高浓度的还原型谷胱甘肽(GSH),发挥解毒功能,使蛋白质处于还原状态[12],维持细胞膜的完整性。当活性氧类(ROS)水平明显升高和(或)氧化还原酶系统受损时,蛋白质发生氧化或蛋白质与氧化还原分子相互作用发生改变,最终可导致细胞的生理功能改变。虽然目前已明确氧化还原酶系统的一项重要功能是参与细胞的自我修复,以保护细胞蛋白不受氧化损伤,但其调节心脏电生理表型的分子机制仍不明确。因此,本研究利用DM大鼠模型探寻Trx系统调节Kv通道重构的机制,结果表明,在DM心肌中,细胞凋亡信号调节通路中JNK是Trx-1的重要结合物,而持续激活的JNK可降低Kv通道表达。
JNK信号通路是丝裂原活化蛋白激酶(MAPK)信号通路的成员之一,它是细胞外界信号由细胞表面转导到细胞核内部的重要传递者[13]。大量的基础研究证实,JNK信号通路参与了细胞增殖与分化、细胞形态维持、细胞骨架构建、细胞凋亡和细胞恶变等多种生物学反应,主要通过JNK磷酸化蛋白的表达来调控相关反应[14]。高糖环境可诱导JNK1磷酸化,进而影响下游TRPC6信号表达,使胶原蛋白沉积过多,导致心肌细胞肥大和纤维化[15]。SP600125是ATP竞争性的JNK高选择性抑制剂,可作用于JNK1、JNK2和JNK3,明显抑制JNK介导的MAPK激活。
应激激活的蛋白激酶(如JNK)是心肌肥厚及凋亡的ROS敏感调节因子[16]。DM状态下JNK会受到刺激进而被激活,而这种激活可通过抗氧化治疗而被抑制[6]。本研究结果显示,这些激酶与心室Kv通道重构有关,DM心肌经过JNK抑制剂处理几个小时后,可以增加Ito密度。
研究证实Kv电流(尤其是Ito)强度降低是多种疾病状态下心室电重构的标志[17],且在大鼠心衰模型中氧化应激和氧化还原酶活性损伤在Kv4通道表达下降及Ito密度降低中具有关键作用[11-12]。本实验室已证实在1型糖尿病[6]或慢性心肌梗死[3]中,Trx系统对调节Kv4通道及Ito密度具有重要作用。
本实验室检测到在糖尿病中心室电重构的标志Ito密度下降可以恢复[3],这一过程称为去重构。在DM心肌中,JNK激酶可以一种TrxR依赖性的方式激活去重构。这些结果表明JNK信号通路可激活DM心肌中的Trx系统,且本研究进一步证实这种激活与TrxR相关,因为JNK激酶抑制剂明显增加Ito通道电流的作用能被TrxR抑制剂所阻断。
现有资料表明,ASK1是慢性心衰中Ito重构的另一关键性氧化还原敏感调节因子[18-19]。ASK1是一种促分裂原活化蛋白激酶,可以激活JNK并受氧化应激特异性调节,ASK1可以与还原型Trx1直接结合,这会降低ASK1的活性[20]。在某些细胞中还原型Grx1也可以与ASK1结合,进而降低ASK1活性。这可能是由于在ROS条件下,Trx1发生氧化,进而释放ASK1,因此ASK1活性增强,对JNK的磷酸化作用也随之增强[5-6]。本研究结果表明,在DM心肌中,Kv4.2通道表达降低与由ASK1所介导的JNK活性增强相关。JNK抑制剂并未明显改变对照组心肌Kv4.2蛋白的丰富度,这也与先前所观察的电生理改变一致。
总之,DM对大鼠心肌细胞的Ito密度有明显影响。DM大鼠左室心肌细胞Trx系统功能下降,蛋白质氧化水平增高,使心肌细胞Ito密度明显下降,这些是导致心室功能降低和心律失常发生的电生理基础,而胰岛素能够逆转DM大鼠心肌细胞Ito的下降[3]。本研究结果表明,Kv通道受氧化还原状态调节,慢性Trx系统损伤可以通过持续激活ASK1-JNK信号通路而促进Ito电流重构。在DM心肌中,IGF-1可激活受体酪氨酸激酶信号通路,进而抑制ASK1-JNK活性,并通过一种TrxR依赖性途径增加Kv4通道。因此,对于由心电重构及DM所致的心律失常,Trx系统可能是一个新的治疗靶点。
【参考文献】
[1]Li XY, Liu G, Guo JH. Research progress of changes of potassium channels in diabetic myocardium[J]. Chin J Cardiac Pacing Electrophysiol, 2012, 26(3): 261-264. [李学永, 刘刚, 郭继鸿.糖尿病心肌钾离子通道改变研究进展[J]. 中国心脏起搏与心电生理杂志, 2012, 26(3): 261-264.]
[2]Veit F, Pak O, Brandes RP. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: focus on ion channels[J]. Antioxid Redox Signal, 2015, 22(6): 537-552.
[3]Zheng MQ, Li XY, Liu G,et al. Role of thioredoxin system to remodeling in diabetes rat hearts[J]. J Clin Cardiol, 2013, 29(8):573-576. [郑明奇, 李学永, 刘刚, 等. 硫氧还蛋白系统对糖尿病大鼠心肌Ito电流的影响[J]. 临床心血管病杂志, 2013,29(8): 573-576.]
[4]Li X, Tang K, Xie B,et al. Regulation of Kv4 channel expression in failing rat heart by the thioredoxin system[J]. Am J Physiol Heart Circ Physiol, 2008, 295(1): H416- H424.
[5]Nicholson CK, Lambert JP, Molkentin JD,et al. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure[J]. Arterioscler Thromb Vasc Biol, 2013,33(4): 744-751.
[6]Zschauer TC, Matsushima S, Altschmied J,et al. Interacting with thioredoxin-1-disease or no disease?[J]. Antioxid Redox Signal,2013, 18(9): 1053-1062.
[7]Liu YN, Xu QL, Guo XH,et al. Effects of MAPKs signaling on heat stress-induced apoptosis of pulmonary microvascular endothelial cells and its mechanism[J]. Med J Chin PLA, 2017,42(4): 279-284. [刘亚楠, 徐秋林, 郭晓华, 等. MAPKs家族在中暑小鼠肺微血管内皮细胞凋亡中的作用及机制研究[J].解放军医学杂志, 2017, 42(4): 279-284.]
[8]Karczewski KJ, Snyder M, Altman RB,et al. Coherent functional modules improve transcription factor target identification,cooperativity prediction, and disease association[J]. PLoS Genet, 2014, 10(2): e1004122.
[9]Ouedraogo R, Wu X, Xu SQ,et al. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells evidence for involvement of a cAMP signaling pathway[J]. Diabetes, 2006, 55(6): 1840-1846.
[10]Adams V, Linke A, Kränkel N,et al. Impact of regular physical activity on the NAD(P)H oxidase and angiotensin receptor system in patients with coronary artery disease[J]. Circulation,2005, 111(5): 555-562.
[11]Li S, Zheng MQ, Rozanski GJ. Glutathione homeostasis in ventricular myocytes from rat hearts with chronic myocardial infarction[J]. Exp Physiol, 2009, 94(7): 815-824.
[12]Zheng MQ, Tang K, Zimmerman MC,et al. Role of gammaglutamyl transpeptidase in redox regulation of K+channel remodeling in postmyocardial infarction rat hearts[J]. Am J Physiol Cell Physiol, 2009, 297(2): C253-C262.
[13]Goolsby TV, Lombardo FA. Extravasation of chemotherapeutic agents: prevention and treatment[J]. Semin Oncol, 2006, 33(1):139-143.
[14]Prakasam A, Ghose S, Oleinik NV,et al. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1[J].Cell Death Dis, 2014, 5: e1358.
[15]Chen XM, Liu XL, Wu W. Relationship between c-junN terminal kinase 1 and early injury in the myocardium of diabetic rats[J].Chin Gen Pract, 2016, 19(27): 3281-3285. [陈昕明, 刘晓亮, 吴伟. c-junN端蛋白激酶1磷酸化与糖尿病大鼠早期心肌损伤关系研究[J]. 中国全科医学, 2016, 19(27): 3281-3285.]
[16]Liang H, Li X, Li S,et al. Oxidoreductase regulation of Kv currents in rat ventricle[J]. J Mol Cell Cardiol, 2008, 44(6):1062-1071.
[17]Kaprielian R, Wickenden AD, Kassiri Z,et al. Relationship between K+channel down - regulation and Ca2+in rat ventricular myocytes following myocardial infarction[J]. J Physiol, 1999,517(1): 229-245.
[18]Nishida K, Otsu K. The role of apoptosis signal-regulating kinase 1 in cardiomyocyte apoptosis[J]. Antioxid Redox Signal, 2006,8(9-10): 1729-1736.
[19]Izumiya Y, Kim S, Izumi Y,et al. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin Ⅱ-induced cardiac hypertrophy and remodeling[J]. Circ Res, 2003, 93(9): 874-883.
[20]Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activityindependent manner[J]. Circ Res, 2002, 90(12): 1259-1266.
