ldenti fication of miRNAs and target genes regulating catechin biosynthesis in tea (Camellia sinensis)
2018-05-08SUNPingZHANGZhenluZHUQiufangZHANGGuoyingXlANGPingLlNYulingLAlZhongxiongLlNJinke
SUN Ping, ZHANG Zhen-lu, ZHU Qiu-fang, ZHANG Guo-ying, XlANG Ping, LlN Yu-ling, LAl Zhongxiong, LlN Jin-ke,
1 College of Horticulture, Fujian Agriculture and Forestry University, Fuzhou 350002, P.R.China
2 Anxi College of Tea Science, Fujian Agriculture and Forestry University, Fuzhou 350002, P.R.China
3 College of Protection, Fujian Agriculture and Forestry University, Fuzhou 350002, P.R.China
1. lntroduction
MicroRNAs (miRNAs) are a class of non-protein-coding RNAs that are 20–24 nucleotides (nt) in length, and have multiple biological functions including negative regulatory roles in gene expression (Rhoadeset al. 2002; Yaoet al.2007). miRNAs are widely distributed in animals, plants,and even viruses, where they play critical roles in complicated biological processes by regulating target mRNAs (Lim and Bartel 2003). In plants, ample evidence exists that miRNAs are involved in many biological processes, including morphogenesis of tissues and organs such as roots,stems, leaves, and flowers (ZhangWet al. 2006; Wang and Fan 2007); response to hormones and environmental stresses (Xu and Xie 2010; Dinget al. 2011) and regulation of floral development (Puzey and Kramer 2009). To date,miRNAs have mainly been predicted and identified using approaches such as high-throughput sequencing, cloning,bioinformatics prediction, and microarray analysis. In this study, we used a bioinformatics approach in conjunction with mature sequences of conserved miRNAs from different plant species to predict potential conserved miRNAs in tea(Camellia sinensis) strain 1005. In addition, we eliminated false positive sequences according to specific characteristics, such as miRNA conservation and length; miRNA precursor hairpin structures and secondary-structure minimum free energies; and mismatches between miRNAs and base complementary sequences of miRNA precursors. Such a bioinformatics-based approach has been widely used to predict and identify multiple miRNAs in many plant species,including 188 miRNAs inZea mays(ZhangWet al. 2006)and conserved miRNAs inCoffea arabica(Akteret al.2014)andGossypium hirsutum(Chenget al.2007).
Tea, which is cultivated extensively worldwide, is economically important because it is one of the world’s major beverages (Linet al. 2003; Mondalet al. 2004). Catechin,an important secondary metabolite in tea, is one of the critical functional components affecting tea quality and flavor. Multiple studies (Giménez 2006; Wanget al. 2012)have demonstrated that catechin has many health effects in humans, such as scavenging free radicals and conferring resistance to oxidation and cancer (Pang and Dixon 2007).To date, studies about regulation of catechin biosynthesis have focused on transcription factors and environmental factors. For example, catechin accumulation is characterized by geography and seasonality, which suggests that catechin biosynthesis is regulated by environmental factors such as temperature and light. Shading in summer has been reported to decrease the catechin content of tea(Zhanget al. 2004), and catechin accumulation was higher in tea calluses cultivated under light (Wanget al. 2008).Researchers have demonstrated that dark treatment reduces tea catechin content; in particular, expression levels of anthocyanidin synthase (ANS), which is involved in catechin biosynthesis, are lowered, while those of chalcone synthase(CHS), flavanone 3-hydroxylase (F3H) and dihydro flavonol 4-reductase (DFR), also involved in the catechin synthesis pathway, remain stable (Honget al. 2014). Some researchers have also reported that short exposure to low-intensity ultraviolet (UV) light can boost the catechin content of tea,whereas prolonged exposure decreases catechin accumulation (Zhenget al. 2010). Catechin content is also affected by water and fertilizer conditions (Yueet al. 2011), carbon supply, and hormones (Wanget al. 2008).
Despite these findings, elucidation of transcriptional and post-transcriptional regulation of catechin synthesis requires further investigation. Transcriptional factors, such asMYB,BHLHandWRKY, and regulatory proteins, have been shown to affect catechin accumulation by regulating expression of genes involved in catechin synthesis (Taylor and Grotewold 2005; Ravagliaet al. 2013). Given the important roles of miRNAs in post-transcriptional regulation of gene expression, elucidation of their involvement in the regulation of catechin synthesis pathway-related gene expression requires further work. In this study, using a bioinformatics prediction method, we aimed to predict and identify miRNAs in tea to determine their post-transcriptional regulatory roles in the expression of genes involved in catechin synthesis.
2. Materials and methods
2.1. Plant materials, miRNAs and nucleotide sequences
All plant materials were obtained from tea strain 1005, a tea germplasm resource with high levels of gallated catechin (Linet al. 2005). First, the third and oldest leaves of tea strain 1005 were harvested in May 2015 for use in a quantitative real-time PCR (qRT-PCR) analysis to test gene and miRNA expression levels. Leaves of tea strain 1005 at different maturities were combined and sent to Novogene (Beijing,China) for construction of a transcriptome library to predict conserved novel miRNAs of tea. Sequences of catechin synthesis-related genes of tea were obtained from the Gen-Bank database (https://www.ncbi.nlm.nih.gov/genbank/) of the National Center for Biotechnology Information (NCBI).Sequences of miRNAs were acquired from miRBase for taxa such asArabidopsis thaliana,Physcomitrella patens,Z.mays,Oryza sativa,Medicagosp.,Glycine max, andVitis vinifera.
2.2. ldenti fication of valid miRNA precursors
To identify valid miRNA precursors, we first aligned the miRNA sequences of other plant species with the transcriptome library of tea strain 1005 and selected miRNAs with three or fewer mismatches that aligned with known miRNAs.We then filtered out protein-coding sequences using NCBI BLASTX. Finally, we analyzed secondary structuresviaMfold (http://unafold.rna.albany.edu/?q=mfold) and chose valid miRNA precursors according to the following criteria(ZhangBet al. 2006a): 1) miRNA precursor sequences had three or fewer mismatches with conserved miRNA sequences from other plants; 2) precursor sequences could form hairpin structures; 3) miRNA sequences were located on one arm of the hairpin structure; 4) miRNA sequences had no more than four mismatches with base complementary sequences of miRNA precursors, which do not have hairpin structures; 5) the minimal free energy of the precursor’s secondary structure was less than −18 kcal mol−1; and 6)the percentage of (A+U) in miRNA sequences was 40–70%.
2.3. Analysis of miRNA levels
Extraction of total miRNAs of tea and synthesis of first-strand cDNAsExtraction of total RNAs from tea was performed using Trizol reagent (Invitrogen, USA) according to the manufacturer’s specifications. Extracted miRNA purity was determined by measuring OD260to OD280ratios on a spectrometer, and integrity was tested by 1% agarose gel electrophoresis. First-strand cDNAs were synthesized using a Mir-XTMmiRNA First-Strand Synthesis and SYBR qRT-PCR Kit (Clontech, USA) following the instructions supplied with the kit.
qRT-PCR analysis of miRNAsspecific primers for miRNA qRT-PCR analysis were designed using DNAMAN Software(Table 1), withU6used as a house-keeping gene (Jeyarajet al. 2014). All data were generated on a Roche LightCycler 480 qRT-PCR instrument (Roche, Switzerland). Excel 2003 was used for data analysis and figures were generated with Graphpad Software.
2.4. Prediction and Identification of miRNAs involved in catechin synthesis
Online prediction of miRNAs interacting with catechin synthesis-related genesThe online software tool psRNATarget (http://plantgrn.noble.org/psRNATarget/) was used to predict miRNAs involved in regulating catechin synthesis-related genes. Target genes were then predicted from the generated scores as follows. First, mismatch scores with potential targets were calculated for miRNAs that were 20 nt in length, with G-U mismatches, deletion/insertion,and other mismatches scored as 0.5, 2 and 1, respectively.Scores for miRNAs with more than 20 nt were obtained by choosing the minimum score calculated from all possible sets of 20 consecutive nucleotides. If a mismatch other than G-U was found in the 2–7 nt region of the 5-terminus of the miRNA under consideration, 0.5 was added to the total score. Mismatches in the middle of the miRNA-mRNA complementary region were acceptable as long as they were located between nucleotides 9 and 11. Finally, sequences with scores no higher than 5 were considered as potential miRNA targets (Table 2).
Veri fication of miRNA cleavage sites in targets by RNA ligase-mediated rapid ampli fication of cDNA ends (RLMRACE)Total RNAs of tea leaves of different maturities and young stems were separately extracted using Trizol reagent(Invitrogen) according to the kit protocol. RNA purity and integrity were tested by 1% agarose gel electrophoresis after measuring RNA concentrations. Equal amounts ofRNA from shoots, leaves of different ages, and young stems were combined and used as a template for RLM-RACE reverse-transcription performed with a GeneRacer Kit (Invitrogen) according to the method of Lin (2011). specific and universal primers designed with DNAMAN Software(Table 3) were used for nested-PCR ampli fication, with the resulting products examined by 1% agarose gel electrophoresis. Target bands were recovered, cloned into vectors, and sent to Huada Biotech (Shenzhen, China) for sequencing to verify cleavage sites on target genes.

Table 1 specific primers used for quantitative real-time PCR(qRT-PCR) of microRNAs (miRNAs) from bioinformatics analysis
2.5. qRT-PCR analysis of miRNAs and target genes
Total RNA was extracted as described above and used as a template to generate first-strand cDNA by a First-Strand Synthesis and SYBR qRT-PCR Kit (TaKaRa, Japan).GAPDH,18S, andβ-actinwere used as house-keeping genes following the method of Sunet al. (2010). All primers were designed using DNAMAN Software and synthesized by Huada Biotech, China (Table 3). qRT-PCR ampli ficationswere carried out in 20-μL reaction volumes consisting of 10 μL SYBR Mix, 0.8 μL each of sense and antisense primers,1 μL cDNA as a template, and 7.4 μL distilled deionized water. Cycling conditions were as follows: initial denaturation at 95°C for 30 s, followed by 45 cycles of denaturation at 95°C for 30 s, annealing for 10 s and extension at 72°C for 15 s, with a melting curve generated at the end of the reaction. All data were obtained on a Roche LightCycler 480 instrument and analyzed in Excel 2003.

Table 2 Potential microRNAs (miRNAs) regulating catechin biosynthetic pathway genes
3. Results
3.1. Prediction of miRNAs in tea and sequence analysis of their precursors
In plants, miRNA sequences are highly conserved. In this study, we used known miRNAs of other plant species from miBase to identify 92 members of 81 miRNA families in tea strain 1005 by high-throughput sequencing of a cDNA library.miRNA sequence lengths ranged from 19 to 24 nt (Appendix A).Although most of the identified miRNA families in tea strain 1005 had only one member, families csn-miR156, csnmiR1863, csn-miR1533, csn-miR1436, csn-miR5998, and csn-miR529 were represented by four, three, three, three,two, and two members, respectively. According to the results of the analysis, only eight miRNAs (miR156j, miR1155,miR2119, miR2925, miR3521, miR399a, miR408-5p, and miR5385) contained low percentages of (A+U) ranging between 40.4 and 48.3%. The (A+U) percentage of the majority of identified miRNAs from tea strain 1005 ranged between 50 and 82.5%, thus satisfying the requirement that the (A+U) percentage should be greater than that of (G+C)in miRNA sequences (Zhang Wet al.2006).
In contrast to animal cells, where the length of mature miRNA precursors ranges from 70 to 80 bp, the length and structure of plant miRNA precursors is highly variable. As shown in Appendix A, lengths of miRNA precursors in tea strain 1005 ranged from 54 to 298 bp. We also found that 47 and 49 miRNAs were located at the 3´ and 5´ ends of the precursor, respectively. Even in miRNAs from the same family, such as miR1436, miR156, miR1863, and miR1533,precursor lengths and locations were variable, further verifying the diversity of the identified miRNA precursors in tea.Characteristics used for bioinformatic prediction of miRNAs include precursor hairpin structure and minimal free energy for folding (Zhang Wet al.2006). miRNA precursors in plants such asArabidopsisare characterized by highly diverse hairpin structures (Bartel 2004; Millar and Waterhouse 2005). In this study, secondary structures of miRNA precursors were analyzed with Mfold Software (Appendix B), which revealed free energies for folding of −9.4 to −80.0 kcal mol−1.This wide range of values may have been due to the great difference in lengths of the miRNA precursors. After their alignment with precursor sequences of other plant speciesfrom miBase, we found that miRNA precursor lengths and stem-loop structures varied among plant species, whereas the locations of mature miRNAs and base complementary sequences of miRNA precursors were conserved.
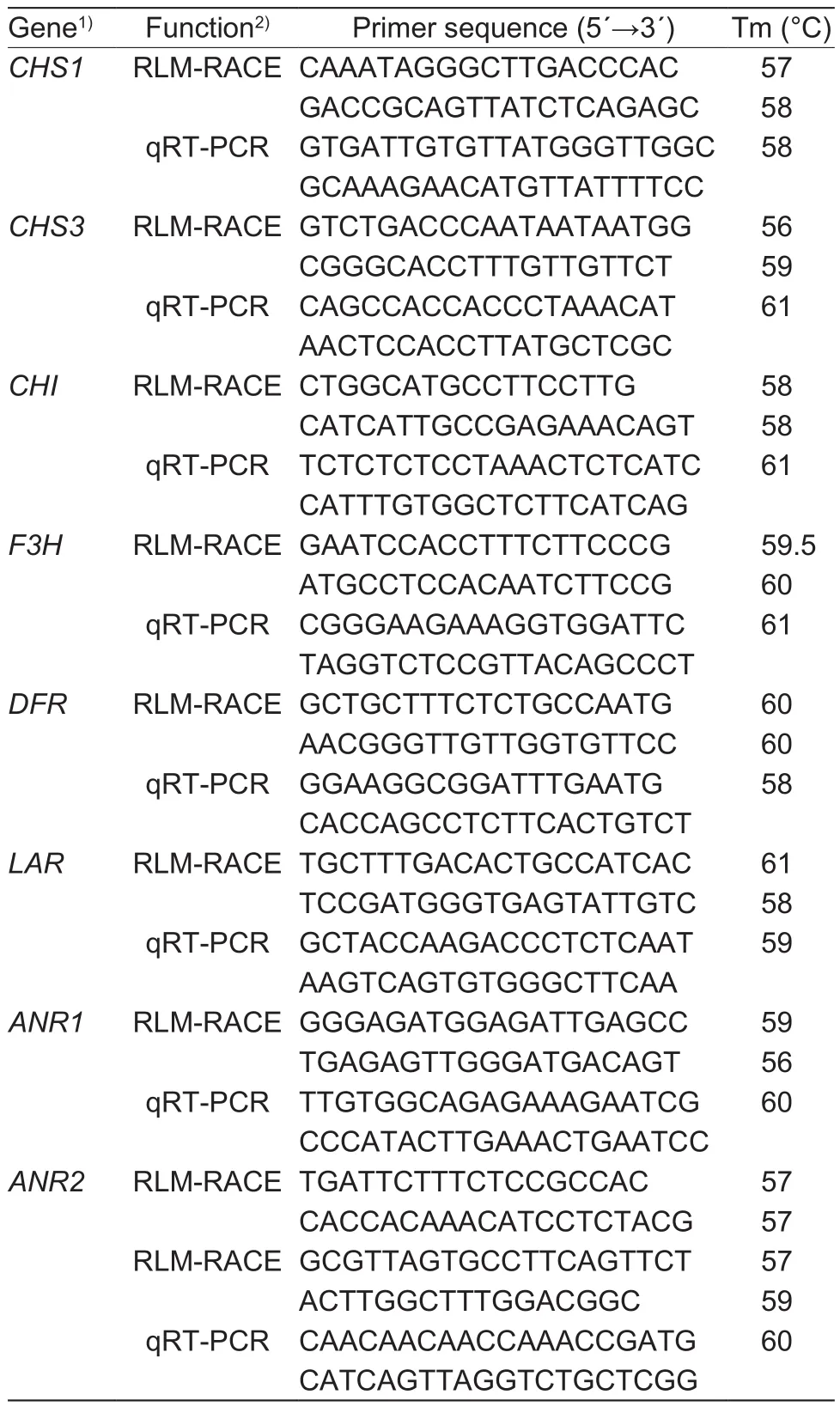
Table 3 Primers used to amplify microRNA (miRNA) cleavage sites
3.2. Expression analysis of identified miRNAs by qRT-PCR
We used qRT-PCR to con firm the existence of miRNAs predicted by the bioinformatics approach in tea strain 1005,thereby verifying the reliability of this method. In addition,we uncovered five expression patterns among the 31 miRNAs analyzed in the qRT-PCR assay (Fig. 1). First, eight miRNAs displayed expression levels that were negatively correlated with tea leaf maturity; in particular, miR1533,miR1863a, miR865-3p, miR399a, miR156g-3p, miR477c,miR5638a, and miR5641 all had higher expression levels in the first leaves compared to the third leaves, but low or even no expression in old leaves. Second, expression levels of another 18 miRNAs (miR156h, miR1863b, miR529-5p,miR164b-3p, miR535, miR5559-5p, miR5021a, miR529d,miR156i, miR2868, miR5240, miR3437-3p, miR5264,miR5180b, miR5385, miR6191, miR6462a, and miR1109)were not obviously correlated with tea leaf maturity. The third leaves generally exhibited the highest expression, followed by the first one, with old leaves having only low or even no expression. miR529d, miRNA1863b, miRNA5021a and miR1109 were exceptions, however, exhibiting similar levels in both first and old leaves. The third expression pattern was exactly contrary to the first one, with low expression levels observed in young leaves that increased with leaf maturation. This expression pattern was only detected for miR3513-3p. Fourthly, miR5998a, miR7814, and miR6261 had the same expression pattern: high expression in first and third tea leaves, but low or even no expressions in old ones. Finally, the expression of miR7539 was steady across all leaf ages, which indicated that miR7539 might play an important role in growth of tea leaves. We hypothesized that miR7539 could be analyzed as a potential reference gene to analyze the quantitative expressions of miRNAs in tea.
3.3. Prediction of miRNA target genes
Using the miRNAs identified in this study and transcriptome sequences of tea strain 1005, we applied psRNATarget Software to predict target genes according to studies of Zhang Wet al.(2006). To annotate these targets, we further aligned them to the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases.After screening out unannotated and repeated sequences,we obtained target genes that can be regulated by miRNAs(Appendix C).
The identified targets were found to be involved in multiple processes, such as plant growth and development and primary and secondary metabolism. According to the results of the KEGG analysis, we arti ficially divided these targets into three classes. The first class included genes for transcriptional regulatory factors involved in plant growth and development. Members of the second class encoded functional proteins related to: starch, sucrose, amino acids,various metabolic processes (such as glycolysis, photosynthesis, the citric acid cycle, oxidative phosphorylation,purine and nitrogen metabolism), carotenoid biosynthesis,and ubiquitin-mediated protein degradation. The third class is comprised of targets involved in biological regulation and interaction with pathogens.

Fig. 1 Expression of microRNAs (miRNAs) of tea leaves with different maturity levels. L1, L3 and L indicate the first, the third and the old leaves in tea 1005 strain, repectively.
3.4. Prediction and Identification of miRNAs involved in regulating catechin biosynthesis-related genes
Catechin, one of the main secondary metabolites in tea plants, is a polyphenol. In tea, both the phenylpropanoid and flavonoid pathways participate in catechin biosynthesis in a manner similar to the anthocyanin biosynthetic pathway operating in plant species such asV.vinifera(Wanget al.2009). In particular, non-galloylated catechins are first produced in the phenylpropanoid and flavonoid pathways and are then converted to ester catechins. To date, multiple key genes have been identified that are related to the phenylpropanoid and flavonoid pathways. To understand the regulatory roles of miRNAs in catechin biosynthesis, we used the identified miRNAs as probes to predict catechin synthesis-related genes that can act as targets, and thereby identified 11 potential targets.
To con firm the predicted results, we next used RLMRACE to detect the cleavage sites of these targets, and thereby con firmed seven targets (Fig. 2).CHS1(D26593.1)andCHS3(D26595.1), which belong to the same family and have highly similar coding regions, were identically cleaved by miR7814. This finding indicates that a single miRNA can target different members of one family at similar or identical cleavage sites. In addition, we found thatANR1andANR2can be cleaved by miR5559-5p at different sites; however,ANR2can also be regulated by miR5264 at multiple cleavage sites, which indicates that a single target gene can be regulated by different miRNAs at various sites. Furthermore, as shown in Fig. 3,CHIandF3Hcan be targeted by miR529d and miR156g-3p at one site each, respectively,whileLARcan be cleaved by miR2868 at two sites. We also observed multiple cleavage sites in a single transcript,and noted that some other sites might be inconsistent with predicted ones. This latter situation may have arisen due to universal cleavage of mRNAs by miRNAs (Kasschauet al.2003) due to small RNA interference (Elbashiret al. 2001).Alternatively, these genes may be regulated simultaneously by some unknown miRNAs or siRNAs. These data reveal the complexity of post-transcriptional regulation of genes and indicate that additional efforts are required to explore miRNAs in tea plants.

Fig. 2 The cleavage sites of target genes by RNA ligasemediated rapid ampli fication of cDNA ends (RLM-RACE).CHS, chalcone synthase; CHI, chalcone isomerase; F3H,flavanone 3-hydroxylase; F35H, flavonoid 3´,5´-hydroxylase;DFR, dihydro flavonol 4-reductase; LAR, leucoanthocyanidin reductase; ANS, anthocyanidin synthase; ANR, anthocyanidin reductase. The boxes indicate the cleavage sites; the numbers indicate the fraction of cloned PCR products terminating at different positions.
3.5. Expression analysis of miRNAs and their targets in tea leaves
To further understand the regulatory roles of the aforementioned miRNAs in target gene expression and catechin synthesis, we used qRT-PCR to monitor the expression of miRNAs and their targets in tea leaves of different ages.The results of this analysis are shown in Fig. 3.
The expression of some miRNAs and their targets were negatively related in tested tea leaves of different ages.The expression patterns of these elements, which included miR529d and its target geneCHI, and miR2868 and its target geneLAR, indicated that the targets involved in catechin synthesis were downregulated by the corresponding miRNAs. We also found that the expression of some miRNAs was negatively related to targets only in certain leaves; for example, expression levels of miR156g-3p were negatively correlated with its targetF3Hin first and third leaves, whereas both had low expression levels in old leaves. Similarly,miR5240 and its target geneDFRwere negatively correlated in all tested leaves except for old ones, where both were not expressed. In addition, expression patterns of some miRNAs and their targets were similar in different leaves or under various treatments. For example, miR7814 was expressed similarly in the first and third leaves, but showed no expression in old leaves. The targets of miR7814, includingCHS1andCHS3, had similar expression patterns,but neither target was negatively correlated with miR7814 in terms of expression levels. Furthermore, the expression pattern of miR5559-5p, which cleaved bothANR1andANR2, did not have an opposite trend compared to its targets; the same was true for miR5246, which targetedANR2as well.
4. Discussion
4.1. Bioinformatics-based prediction of miRNAs
Much evidence currently exists that miRNAs play essential regulatory roles in many aspects of plant growth and development, including nutrition and secondary metabolism,organelle morphogenesis, and response to environmental stresses. At present, the main approaches used to isolate and identify miRNAs from plants are direct cloning,Northern blotting,in situhybridization, microarray analysis,high-throughput sequencing, and bioinformatics-based prediction. The latter strategy was the main approach used in this study to identify miRNAs from tea strain 1005. Bioinformatics prediction of novel miRNAs is performed computationally based on the conservation of known miRNAs and secondary structural features of plant miRNA precursors,including minimum folding free energy index (MFEI) and mismatches between miRNAs and base complementary sequences of miRNA precursors (Zhanget al. 2007). This approach has been widely used to identify novel miRNAs in many plant species, such asV.vinifera(Songet al. 2010b),Gossypiumspp.(Qiuet al. 2007),B.campestris(Xieet al.2007),Z.mays(Zhang Bet al. 2006), andC. arabica(Deviet al. 2016). Furthermore, miRNAs predicted by the bioinformatics approach have been subsequently con firmed by qRT-PCR, high-throughput sequencing, and various other methods (Xieet al.2007; Songet al. 2010a). The bioinformatics method has been used to identify novel miRNAs in tea (Prabu and Mandal 2010); for example, 13 conserved miRNAs in nine families were detected from a tea expressed sequence tag database (Das 2010).
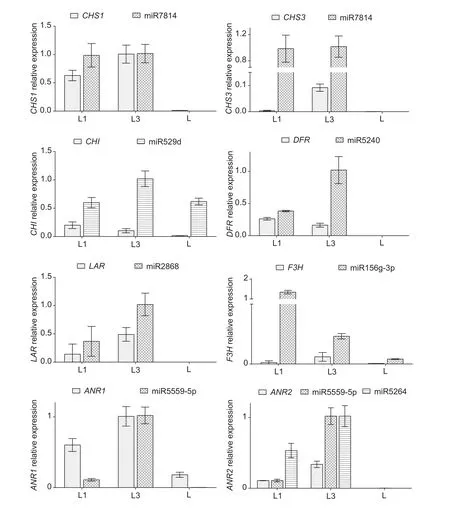
Fig. 3 Analysis of expression of microRNA (miRNA) and target gene in different tea leaves. CHS, chalcone synthase; CHI,chalcone isomerase; DFR, dihydro flavonol 4-reductase; LAR, leucoanthocyanidin reductase; F3H, flavanone 3-hydroxylase; ANR,anthocyanidin reductase. The x axis indicated different tea leaves; and L1, L3, and L indicated the first, third, and the old leaves in tea 1005 strain, respectively. And the errors bars indicated SD.
In this study, we identified 55 conserved miRNAs based on known miRNAs and using a transcriptome database of tea strain 1005. We used qRT-PCR to con firm this result and found 25 miRNAs expressed in tea leaves of different ages. The possible reason for this discrepancy may be that some miRNAs were low-copy or weakly expressed;however, some miRNAs could not be detected by qRT-PCR.Another possible reason is lack of primer specificity due to the unavailability of exact sequences of miRNAs predicted by the bioinformatics method, which may have hindered detection as well. The rest of the miRNAs could be verified by cloning or sequencing in future experiments.
4.2. The important roles of miRNAs in catechin biosynthesis
The health bene fits of catechin, the main polyphenol and secondary metabolite in tea, include antioxidant, anti-diabetes, anti-cardiovascular disease, and even anticancer activities (Giménez 2006; Wanget al. 2012). To further understand the function of miRNAs in regulating catechin biosynthesis, we used RLM-RACE to identify eight miRNAs targeting seven genes involved in catechin synthesis. Our findings demonstrate that these miRNAs play negative regulatory roles during catechin synthesis by cleaving target genes.
CHS is a rate-limiting enzyme inflavonoid and isoflavone synthesis. Tea plants possess fourCHSgenes, three of which,CHS1,CHS2, andCHS3, are homologous (Takeuchiet al. 1994). Our RLM-RACE experiments con firmed thatCHS1andCHS3are simultaneously cleaved by miR7814.Zhanget al. (2014) have reported thatCHS1andCHS3are structurally conserved and that no intron is present in theCHS2DNA sequence. We thus speculate that the factors transcriptionally regulatingCHS2are different from those controllingCHS1andCHS3. In addition, we experimentally verified that miR529d can cleave the target geneCHIbecause the expression of miR529d and theCHIgene in tea leaves of different ages showed opposite trends, further con firming that miR529d is a negative regulatory factor of theCHIgene. F3H is a key enzyme in the catechin biosynthesis pathway (Punyasiriet al. 2004). A previous study has revealed that expression of theF3Hgene is down-regulated under abscisic acid treatment (Singhet al. 2008). Moreover, enzyme activities of F3H and DFR are improved after pre-harvest spraying with thidiazuron, a cytokinin activator(Moalembenoet al. 1997).
DFR, a key enzyme of the catechin biosynthesis pathway,catalyzes the production of the common precursor of catechin, anthocyanin, and procyanidine synthesis pathways(Xia and Gao 2009). Little evidence exists, however, thatDFRexpression can be controlled by miRNAs. One of the few pieces of such evidence is that DFR can be regulated by miR156 through SQUAMOSA promoter-binding protein-like(SPL) transcription factors inArabidopsis(Cuiet al. 2014).Given the fact thatDFRcan also be cleaved by miR156 in tea, miR156 has been predicted to affect the catechin content of tea plants through its regulatory role inDFRexpression (Fanet al. 2015). In addition to DFR, LAR is an important enzyme in catechin biosynthesis (Ma 2007), and both of these enzymes are thought to influence catechin content. Our data revealed thatLARis negatively regulated by miR2868 at the post-transcriptional level because they had reverse expression trends; even thoughDFRcould be cleaved by miR5240, their expression patterns did not show clear correlations.ANR, a multigene family, converts anthocyanin into epicatechin and epigallocatechin. AlthoughANRgenes are widespread in plants, the regulation of their expression has not been well studied. Previous investigations have demonstrated thatANRis regulated by the fifth subgroup of R2R3-MYB and that transgenesis of CsMYB5-2 intoNicotiana benthamianacan significantly promoteANRexpression in flowers of the resulting transgenic plants (Zhao 2013). To date, only twoANRgenes,ANR1andANR2,have been identified in tea. We found that both genes are controlled by miR5559-5p. In addition,ANR2is also regulated by miR5246.
5. Conclusion
In total, we computationally identified 92 conserved miRNAs in tea strain 1005 and verified presence of 31 miRNAs with a qRT-PCR assay. We also identified seven miRNAs(miR7814, miR529d, miR5240, miR5559-5p, miR5264,miR156g-3p, and miR2868) cleaving eight catechin synthesis pathway-related genes, includingCHS,CHI,DFR,ANR,LAR,andF3Hthrough RLM-RACE and qRT-PCR experiments using tea leaves of varying ages. Our data suggest that these miRNAs negatively regulate catechin synthesis by cleaving target genes involved in the catechin biosynthesis pathway.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (31170651), the Project from the Ministry of Agriculture, China (KCa16022A), and the Major Science and Technology Project in Fujian Province, China(2015NZ 0002-1).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
Akter A, Islam M M, Mondal S I, Mahmud Z, Jewel N A,Ferdous S, Amin M R, Rahman M M. 2014. Computational Identification of miRNA and targets from expressed sequence tags of coffee (Coffea arabica).Saudi Journal of Biological Sciences, 21, 3–12.
Bartel D P. 2004. MicroRNAs: Genomics, biogenesis,mechanism, and function.Cell, 116, 281–297.
Cheng X Q, Fu L X, Yi Y Z, Kai G, Si Q H, Li N, Zhi M Y. 2007.Computational Identification of microRNAs and their targets inGossypium hirsutumexpressed sequence tags.Gene,395, 49.
Cui L G, Shan J X, Shi M, Gao J P, Lin H X. 2014. The miR156-spl9-dfr pathway coordinates the relationship between development and abiotic stress tolerance in plants.Plant Journal for Cell & Molecular Biology, 80, 1108–1117.
Das A. 2010. Computational Identification of conserved microRNAs and their targets in tea (Camellia sinensis).American Journal of Plant Sciences, 1, 77–86.
Devi K J, Chakraborty S, Deb B, Rajwanshi R. 2016.Computational Identification and functional annotation of microRNAs and their targets from expressed sequence tags(ESTs) and genome survey sequences (GSSs) of coffee(Coffea arabica).Plant Gene, 6, 30–42.
Ding Y, Zhu C, Wang S, Liu H. 2011. Regulation of miRNAs response to heavy metal stress in plant.Process in Biochemistry and Biophysics, 38, 1106–1110.
Elbashir S M, Harborth J, Lendeckel W, Yalcin A, Weber K,Tuschl T. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in mammalian cell culture.Nature, 411,494-498.
Fan K, Fan D, Ding Z, Su Y, Wang X. 2015.Cs-miR156is involved in the nitrogen form regulation of catechins accumulation in tea plant (Camellia sinensis).Plant Physiology & Biochemistry, 97, 350–360.
Giménez R. 2006. Bene ficial effects of green tea - a review.Journal of the American College of Nutrition, 25, 79–99.
Hong G, Wang J, Zhang Y, Hochstetter D, Zhang S, Pan Y, Shi Y, Xu P, Wang Y. 2014. Biosynthesis of catechin components is differentially regulated indark-treated tea(Camellia sinensis).Plant Physiology & Biochemistry, 78,49–52.
Jeyaraj A, Chandran V, Gajjeraman P. 2014. Differential expression of microRNAs in dormant bud of tea (Camellia sinensis(L.) O. Kuntze).Plant Cell Reports, 33, 1053–1069.
Kasschau K D, Xie Z, Allen E, Llave C, Chapman E J, Krizan K A, Carrington J C. 2003. P1/HC-Pro, a viral suppressor of RNA silencing, interferes withArabidopsisdevelopment and miRNA function.Developmental Cell, 4, 205.
Lim L P, Bartel D P. 2003. Vertebrate microRNA genes.Science, 299, 501.
Lin J, Zheng J, Chen R, Chen C. 2005. Screening specific tea plant germplasm resources (Camellia sinensis) with high egcg content.Acta Agronomica Sinica, 31, 1511–1517.(in Chinese)
Lin Y. 2011. Studies on cloning, expression and regulation ofsodgene family during somatic embryogenesis inDimocarpus longan. Ph D thesis, Fujian Agriculture and Forestry University, Fuzhou. (in Chinese)
Lin Y S, Tsai Y J, Tsay J S, Lin J K. 2003. Factors affecting the levels of tea polyphenols and caffeine in tea leaves.Journal of Agricultural & Food Chemistry, 51, 1864.
Ma C. 2007. Molecular cloning and expression analysis of chalcone isomerase, flavonol synthase and leucoanthocyantin reducase genes of teaplant (Camellia sinensis). MSc thesis, Chinese Academy of Agricultural Sciences, China.(in Chinese)
Millar A A, Waterhouse P M. 2005. Plant and animal microRNAs:Similarities and differences.Functional & Integrative Genomics, 5, 129–135.
Moalembeno D, Tamari G, Leitnerdagan Y, Borochov A, Weiss D. 1997. Sugar-dependent gibberellin-induced chalcone synthase gene expression inPetunia corollas.Plant Physiology, 113, 419–424.
Mondal T K, Bhattacharya A, Laxmikumaran M, Ahuja P S. 2004.Recent advances of tea (Camellia sinensis) biotechnology.Plant Cell Tissue & Organ Culture, 76, 195–254.
Pang Y, Dixon R A. 2007. Early steps in proanthocyanidin biosynthesis in the model legumeMedicago truncatula.Plant Physiology, 145, 601–615.
Prabu G R, Mandal A K A. 2010. Computational Identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis).Genomics,Proteomics &Bioinformatics, 08, 113–121.
Punyasiri P A, Abeysinghe I S, Kumar V, Treutter D, Duy D,Gosch C, Martens S, Forkmann G, Fischer T C. 2004.Flavonoid biosynthesis in the tea plantCamellia sinensis:Properties of enzymes of the prominent epicatechin and catechin pathways.Archives of Biochemistry & Biophysics,431, 22–30.
Puzey J R, Kramer E M. 2009. Identification of conservedAquilegia coeruleamicroRNAs and their targets.Gene,448, 46–56.
Qiu C X, Xie F L, Zhu Y Y, Guo K, Huang S Q, Nie L, Yang Z M. 2007. Computational Identification of microRNAs and their targets inGossypium hirsutumexpressed sequence tags.Comparative & Functional Genomics, 395, 49–61.
Ravaglia D, Espley R V, Henry-Kirk R A, Andreotti C, Ziosi V, Hellens R P, Costa G, Allan A C. 2013. Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors.BMC Plant Biology, 13, 53.
Rhoades M W, Reinhart B J, Lim L P, Burge C B, Bartel B,Bartel D P. 2002. Prediction of plant microRNA targets.Cell, 110, 513–520.
Singh K, Rani A, Kumar S, Sood P, Mahajan M, Yadav S K,Singh B, Ahuja P S. 2008. Singh K, Rani A, Kumar S. An early gene of flavonoid pathway, flavanone 3-hydroxylase,exhibits a positive relationship with catechins content in tea (Camellia sinensis(L.) O. Kuntze).Tree Physiology,28, 1349–1356.
Song C, Fang J, Chen W, Lei G, Nicholas K K, Ma Z. 2010a.MiR-RACE, a new efficient approach to determine the precise sequences of computationally identified trifoliate orange (Poncirus trifoliata) microRNAs.PLoS ONE, 5,e10861.
Song C, Jia Q, Fang J, Li F, Wang C, Zhang Z. 2010b.Computational Identification of citrus microRNAs and target analysis in citrus expressed sequence tags.Plant Biology,12, 927–934.
Sun M L, Wang Y S, Yang D Q, Wei C L, Gao L P, Xia T, Shan Y, Lou Y. 2010. Reference genes for real-time fluorescence quantitative PCR inCamellia sinensis. Chinese Bulletin of Botany, 45, 579–587. (in Chinese)
Takeuchi A, Matsumoto S, Hayatsu M, 1994. Chalcone synthase fromCamellia sinensis: Isolation of the cDNAs and the organ-specific and sugar-responsive expression of the genes.Plant & Cell Physiology, 35, 1011.
Taylor L P, Grotewold E. 2005. Flavonoids as developmental regulators.Current Opinion in Plant Biology, 8, 317–323.
Wang C, Fang J, Cao X, Yang G. 2009. Metabolism of proanthocyanidins in gape.Chinese Agricultural Science Bulletin, 25, 169–173. (in Chinese)
Wang D, Will T E, Wang Y, Wan X, Zhang J. 2012. Encapsulated nanoepigallocatechin-3-gallate and elementalSelenium nanoparticlesas paradigms for nanochemoprevention.International Journal of Nanomedicine, 7, 1711–1721.
Wang L, Fan Y. 2007. Process of microRNA in plant.Journal of Agricultral Science and Technology, 9, 18–23.
Wang R, Zhou W, Jiang X. 2008. Reaction kinetics of degradation and epimerization of epigallocatechin gallate(EGCG) in aqueous system over a wide temperature range.Journal of Agricultural & Food Chemistry, 56, 2694–2701.
Xia T, Gao L. 2009. Advances in biosynthesis pathways and regulation of flavonoids and catechins.Scientia Agricultura Sinica,42, 2899–2908. (in Chinese)
Xie F L, Huang S Q, Guo K, Xiang A L, Zhu Y Y, Nie L, Yang Z M.2007. Computational Identification of novel microRNAs and targets inBrassica napus.FEBS Letters, 581, 1464–1474.
Xu Z, Xie C X. 2010. Advances on plant microRNA and stresses response.Hereditas, 32, 1018–1030.
Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu J K, Sun Q. 2007.Cloning and characterization of microRNAs from wheat(Triticum aestivum).Genome Biology, 8, R96.
Yue M C, Tsao T M, Cheng C L, Lin K C, Ming K W. 2011.Aluminium and nutrients induce changes in the pro files of phenolic substances in tea plants (Camellia sinensis).Journal of the Science of Food & Agriculture, 91, 1111–1117.
Zhang B, Pan X, Anderson T A. 2006a. Identification of 188 conserved maize microRNAs and their targets.FEBSLetters, 580, 3753–3762.
Zhang B, Pan X, Cannon C H, Cobb G P, Anderson T A. 2006b.Conservation and divergence of plant microRNA genes.Plant Journal, 46, 243–259.
Zhang B, Wang Q, Wang K, Pan X, Liu F, Guo T, Cobb G P,Anderson T A. 2007. Identification of cotton microRNAs and their targets.Gene, 397, 26–37.
Zhang L Q, Wei K, Wang L Y, Cheng H, Liu B Y, Gong W Y.2014. The structure and single nucleotide polymorphism analysis of chalcone synthase genes in tea plant (Camellia sinenesis).Scientia Agricultura Sinica,47, 133–144. (in Chinese)
Zhang W, Liang Y, Zhang F, CHeng C, Zhang Y, Cheng R,Weng B. 2004. Effects on the yield and quality of oolong tea by covering with shading net.Journal of Tea Science,24, 276–282. (in Chinese)
Zhang W, Luo Y, Li S. 2006. MicroRNA and its role in plant growth and development.Plant Physiology Communications,42,1015–1020.
Zhao L. 2013. Transcription factors analysis inflavonoid biosynthesis pathway and gene function of aivr in tea plant (Camellia sinensis). Ph D thesis, Anhui Agricultural University, China. (in Chinese)
Zheng X Q, Jin J, Chen H, Du Y Y, Ye J H, Lu J L, Lin C, Dong J J, Sun Q Y, Wu L Y, Liang Y R. 2010. Effect of ultraviolet b irradiation on accumulation of catechins in tea (Camellia sinensis).African Journal of Biotechnology, 7, 3283–3287.
杂志排行

Journal of Integrative Agriculture的其它文章
- Detection and characterization of an isolate of Tomato mottle mosaic virus infecting tomato in China
- Yield and water use responses of winter wheat to irrigation and nitrogen application in the North China Plain
- A simulation of winter wheat crop responses to irrigation management using CERES-Wheat model in the North China Plain
- ldentification of the strain-specifically truncated nonstructural protein 10 of porcine reproductive and respiratory syndrome virus in infected cells
- Effect of dietary supplementation with flavonoid from Scutellaria baicalensis Georgi on growth performance, meat quality and antioxidative ability of broilers
- Variability in total antioxidant capacity, antioxidant leaf pigments and foliage yield of vegetable amaranth