Sotos综合征2例报道及文献复习
2018-05-08陆勇刚胥雨菲姚如恩郁婷婷王秀敏沈亦平
陆勇刚, 胥雨菲, 姚如恩, 李 牛, 郁婷婷, 王秀敏, 沈亦平, 王 剑
(1.上海交通大学医学院附属上海儿童医学中心医学遗传科分子诊断实验室,上海 200127;2.上海交通大学医学院附属上海儿童医学中心内分泌遗传代谢科,上海 200127;3.美国哈佛大学医学院附属波士顿儿童医院基因诊断实验室,美国 波士顿 02115)
Sotos综合征是一类先天性过度生长疾病,临床主要表现为典型的特殊面容:巨颅,前额头突出,眼睑裂下斜,下颌尖长,前发际线高,头颅巨大;青少年期生长过快:身高和头围明显增加,大于同地区、同性别、同龄儿童的第97百分位数;神经心理发育迟缓:语言学习障碍及智力低下;骨龄提前[1]。目前认为该疾病与核受体结合SET域蛋白(nuclear receptor-binding SET-domain-containing protein 1,NSD1)基因异常有关,其定位于5号染色体q35位置,于2002年在1例新生平衡易位(5;8)(q35;q24.1)患者中成功分离出该基因,并与小鼠NSD1基因具有同源性[2-3]。NSD1基因由23个外显子组成,开放阅读框从第2个外显子开始,编码1个由2 696个氨基酸组成的蛋白质。NSD1基因缺失或其基因内突变而导致的单倍剂量不足是Sotos综合征的主要致病原因,约有70%~90%的患者可以检测出NSD1基因的异常,包括无义突变、错义突变、移码突变、剪接突变和大片段缺失等[4-5]。
截至2016年,中国内地可以检索到并进行明确基因诊断的Sotos综合征共3例,且都为5q35缺失型患者[6-7]。但中国香港的病例研究发现,中国香港人群中Sotos综合征NSD1基因突变检出率为72%,其中只有8%的患者是5q35缺失型,其余64%均为基因内突变型[8],两者区别较大。我们报道的2例Sotos综合征病例中,1例为5q35缺失型,另1例为基因内突变型,这是目前中国内地首例无义突变病例,该位点突变未见于中国香港人群报道中。为此,我们通过复习文献并结合这2例病例,探讨不同基因型与表型的相关性,以期提高临床诊断率。
1 材料和方法
1.1 病例资料
病例1:男性,8个月,因“发育落后”于2015年7月16日来上海交通大学附属上海儿童医学中心就诊。足月剖宫产,出身时体重4.1 kg,皮肤轻度黄染。就诊时神经系统发育落后于同龄儿,反应欠灵敏,独坐不稳。体格检查:头围48.5 cm(>3s),头颅大,前囟(2×2)cm,舟状头,前额突出,前顶头部毛发稀疏,眼距宽,眼裂下斜,双手、双足大,右手食指屈曲畸形,皮肤粗糙,四肢肌张力可[图1(a)]。头颅磁共振显示胼胝体发育不良,脑室偏大,脑电图正常。心脏超声示动脉导管未闭。肾脏B超未见明显异常。Gesell评分提示有轻度智力低下。临床拟诊为Sotos综合征。随访至2岁,身高95 cm,体重13 kg,语言表达少,喜欢独处,至今不会独走。
病例2:男性,3岁,因“发育落后”于2016年3月30日来上海交通大学附属上海儿童医学中心就诊。神经系统发育落后,17个月会走路,2岁能叫妈妈,目前语言表达少。有癫痫发作史,曾注射流感疫苗出现抽搐,2岁半时发生低热抽搐,10 d前出现无热抽搐,发作后体温39 ℃。体格检查:身高101 cm,体重17 kg,头围52.8 cm(>2s),头颅大,脸长,前额突出,下颌尖长,前顶头部毛发稀疏,眼距宽,眼裂下斜[图2(a)]。头颅磁共振显示脑室扩张,脑水肿可能。心脏超声示动脉导管未闭。肾脏B超未见明显异常。Gesell评分提示有轻度智力落后,特别语言能和应人能较差,有自闭症倾向。临床拟诊为Sotos综合征。
1.2 检测方法
1.2.1 样本留取方法 经上述患儿监护人知情同意,采患儿外周静脉血2 mL,采用QIAamp DNA Blood Mini试剂盒(德国Qiagen GmbH公司)提取基因组DNA,使用NanoDrop 2000分光光度计(美国Thermo Scientific公司)定量后,- 20 ℃保存。
1.2.2 基因芯片 取250 ng DNA,使用限制性内切酶NspⅠ进行酶切消化。采用CytoScan HD Array试剂盒(美国Affymetrix公司)进行基因芯片杂交,严格按试剂说明书操作。应用Chromosome Analysis Suite(ChAS 3.1)软件(美国Affymetrix公司)进行数据分析。
1.2.3 高通量测序 取3 μg DNA,使用Covarias M220核酸剪切仪(美国Covaris公司)将DNA打断成150~200 bp的片段。根据实验手册,采用SureSelectXT Human All Exon试剂盒(美国Agilent Technologies公司)进行文库构建。构建的文库在Illumina cBot簇生成系统(美国Illumina公司)上进行桥式扩增,之后在HiSeq 3000测序系统(美国Illumina公司)上进行测序。测序数据经过质量评估后,使用NextGENe v 2.4.1软件(美国SoftGenetics公司)将读序列与参考基因组(Human 37.3,SNP135;http://hgdownload.soe.ucsc.edu/goldenPath/hg19/snp135Mask/)进行比对以及SNVs和Indels的标注,结果转换成VCF格式文件上传至Ingenuity Variant Analysis平台(美国Qiagen GmbH公司),对SNVs和Indels进行注释和过滤筛选。如发现有候选致病性变异,再用一代测序验证。
2 结果
2.1 芯片分析结果
病例1患儿采用全基因组芯片分析,发现在5号染色体q35.2-35.3位置有1个杂合性缺失,基因位置为chr5:175,570,677~177,477,711,大小为1 907 kb[图1(b)]。NSD1基因位于5号染色体177,133,025~177,300,215,该区域位于缺失片段内,提示NSD1基因整个丢失。
2.2 基因测序结果
病例2患儿通过高通量测序,发现NSD1基因存在1个杂合无义突变c.1262G>A, p.Trp421*,并经一代测序验证(图2)。父母未发现该基因型变异,因此患儿为新生突变(De novo)。该基因变异位点位于chr5:177,209,661,在第5个外显子上,密码子TGG改变形成1个终止密码子TAG,蛋白质翻译在第421位的色氨酸前提前终止。
2.3 文献阅读分析
通过阅读文献[6-7]并结合本研究的这2例病例,分析了中国内地5例Sotos综合征的临床表现和NSD1基因突变的类型。具体结果见表1。

图1 病例1患儿特殊面容及基因芯片结果

图2 病例2患儿特殊面容及基因测序结果
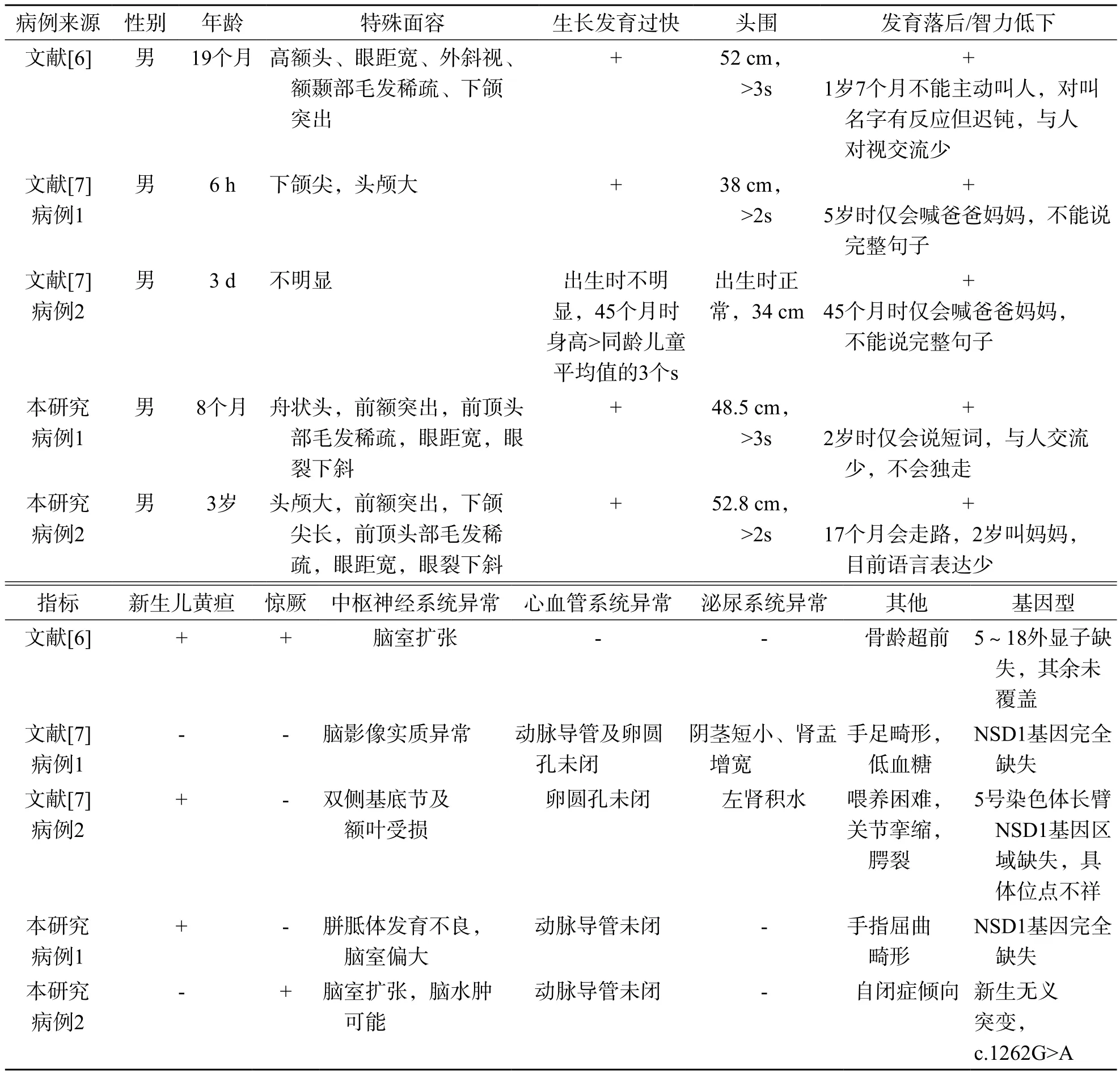
表1 中国内地5例Sotos综合征病例的临床表现和NSD1基因突变类型
3 讨论
中国内地这5例Sotos综合征病例大多表现为生长发育过快,尽管其中1例出生时过度生长不明显,但随访至45个月时身高>同龄儿童平均值的3s,其中3例具有典型的特殊面容,前额突出、前额毛发稀少、眼距宽及眼裂下斜。另2例为新生儿,可能表现不明显。5例病例的共同特征是生长发育过快但表现不一,头颅巨大(1例出生时不明显),具有不同程度的智力低下,尤其语言发育都比较落后,并且中枢神经系统都有影像学的改变,这些表现完全符合Sotos综合征的诊断标准[9]。其他个别的特征还包括骨龄超前、新生儿黄疸、新生儿喂养困难、心血管系统异常、泌尿系统畸形、惊厥、手足畸形等,这些都属于Sotos综合征的次要表现[10]。在成人中,该疾病最常见的症状依次为学习困难、脊柱侧凸、眼科疾病、精神行为失常和大脑影像异常[11]。
这5例病例均检测出了NSD1基因异常,其中4例为5q35缺失型,另1例为基因内突变型。但中国香港地区患者的5q35缺失型检出率仅为12%(3/26),多数为基因内突变型,检出率达88%(23/26)[8]。而在日本患者的研究中,5q35缺失型的比例高达82%(49/60),基因内突变型的比例只有18%(11/60)[12],两者差异明显,这可能跟种族基因结构相关。在日本患者中,其NSD1基因两端侧翼存在低拷贝重复,减数分裂时会发生非等位同源重组,导致NSD1基因缺失,具有相同的断裂位点[12]。由于样本量过小,我们无从推测中国内地的基因突变谱,但鉴于中国香港地区的基因型分布,中国内地的基因内突变型比例可能偏低,这可能跟基因的检测技术相关。目前,中国内地医院临床上基因检测主要采用芯片、多重连接探针扩增等技术,高通量测序技术还没有大量普及,因此无法检测出基因内突变型的患者,漏检的可能性很高。
NSD1蛋白包含许多保守功能域,包括2个PWWP,4个PHD,1个相邻的AWS(Pre-SET)、SET和Post-SET结构域(www.uniprot.org),其中PHD和PWWP是一类解读组蛋白甲基化状态的效应分子,根据组蛋白修饰状态发挥相应的生物学功能或招募其他蛋白共同产生效应[13-14]。另3个SET相关功能域具有组蛋白甲基转移酶活性,主要对核小体组蛋白H3的第36位赖氨酸(H3K36)进行甲基化修饰[15-16]。在4例5q35缺失型患儿中有2例NSD1基因完全缺失,1例明确第5~18外显子缺失,另1例由于技术限制,只知道染色体长臂NSD1基因区域缺失,但不清楚具体位点。还有1例无义突变患儿的蛋白翻译终止在第420位氨基酸,只保留了1个PWWPⅠ功能域,其他功能域缺失。显而易见,这5例病例的NSD1基因结构的杂合性缺失或改变导致一半的NSD1蛋白无法有效表达或蛋白结构遭破坏而无相应的功能,出现了Sotos综合征的表现,是1个剂量敏感型基因。目前认为NSD1是1个辅助转录调节因子,具有转录抑制和转录激活的双向调控作用,对胚胎早期及出生后生长发育起重要作用[17]。Sotos综合征的这种既身体生长过快又精神发育迟缓的矛盾表现很可能与NSD1蛋白的双向调控异常相关。另外,5例病例都有脑影像异常、智力落后、语言学习障碍,提示NSD1对大脑发育有重要影响。
另外,有研究认为Sotos综合征临床表现的严重程度与基因结构的破坏程度呈正相关[10,18-20]:5q35缺失型患者的临床症状最严重,出现严重智力落后、心血管疾病、泌尿系统畸形和中枢神经系统异常的频率更高,还可能跟高胰岛素血症性低血糖症和肿瘤相关,但过度生长表现较轻微;截短突变(无义突变、剪接突变和移码突变)次之,仅表现轻度智力低下,其他系统异常比例较低,然而过度生长表现最严重;错义突变患者临床表现最轻微,智力可正常,也无其他系统病变,只有过度生长表现,并且都发生在蛋白功能域中。但本研究的1例患儿是5q35缺失型,另1例截短突变也可能因为发生在基因头部,导致大部分蛋白功能域缺失,表型与5q35缺失型患儿相差无几,而且这2类基因型样本量过少,并不足以区分截短突变和5q35缺失型的临床特征。到目前为止,我们还未检测到错义突变的患者,不了解其临床表现程度。通过对照基因型与临床表型,我们认为蛋白功能域中的错义突变是研究该功能域具体作用的理想模型。
Sotos综合征的确诊主要基于临床表现和基因诊断。对于临床上典型的或疑似的Sotos综合征患者,首先可采用基因芯片或多重连接探针扩增技术等技术检测染色体拷贝数变异,如未发现任何基因的大片段缺失,必须进一步进行基因测序,检测可能的致病性单核苷酸变异,以免漏检NSD1基因内突变。但目前也只有70%~90%的Sotos综合征患者可检测出NSD1基因异常,剩余10%~30%的患者或由于临床上被错误归入Sotos综合征,或以目前的检测技术水平未能发现NSD1基因变异。CHOUFANI等[21]依据组蛋白修饰与DNA甲基化之间的相互联系,开发了一种表观遗传学诊断方法,通过分析NSD1+/-特异性的CpG甲基化水平诊断Sotos综合征。与正常对照相比,NSD1的功能缺失性突变会导致相关位点的CpG甲基化水平明显下降。该方法的敏感性和特异性均为100%,可以明确诊断Sotos综合征,并可与其他具有交叉临床表现的疾病如Weaver综合征鉴别,还能区分临床意义未明的突变是良性还是致病性的。
目前,针对Sotos综合征患者没有根本性的治疗方法,只能对症治疗,以减轻或预防相关临床症状。随着医疗技术的进步,我们有希望采用基因治疗的手段治愈患者。现今研究最热的技术是CRISPR-Cas9基因编辑系统,对于NSD1基因内突变,在突变位点可引入一条正常的单链DNA模板,利用CRISPR-Cas9打断双链DNA,通过同源重组修复该基因[22]。但对于整个NSD1基因的缺失,应用转基因技术,借助病毒载体在特殊染色体位置整合一个有功能的新基因可能是个更好的选择。
[1]COLE T R,HUGHES H E. Sotos syndrome:a study of the diagnostic criteria and natural history [J].J Med Genet,1994,31(1):20-32.
[2]IMAIZUMI K,KIMURA J,MATSUO M,et al.Sotos syndrome associated with ade novobalanced reciprocal translocation t(5;8)(q35;q24.1)[J]. Am J Med Genet,2002,107(1):58-60.
[3]KUROTAKI N,HARADA N,YOSHIURA K,et al. Molecular characterization of NSD1,a human homologue of the mouse Nsd1 gene [J]. Gene,2001,279(2):197-204.
[4]FARAVELLI F. NSD1 mutations in Sotos syndrome[J]. Am J Med Genet C Semin Med Genet,2005,137C(1):24-31.
[5]KUROTAKI N,IMAIZUMI K,HARADA N,et al. Haploinsufficiency of NSD1 causes Sotos syndrome [J]. Nat Genet,2002,30(4):365-366.
[6]王旭. Sotos综合征NSD1基因缺失突变1例报告并文献复习 [J]. 中国实用儿科杂志,2013,28(6):461-463.
[7]孙碧君,杨琳,梅枚,等. 新生儿Sotos综合征二例报道及文献复习 [J]. 中华围产医学杂志,2015,18(2):127-130.
[8]TONY T M,HAU E W,LO I F,et al. Spectrum ofNSD1 gene mutations in southern Chinese patients with Sotos syndrome [J]. Chin Med J(Engl),2005,118(18):1499-1506.
[9]TATTON-BROWN K,RAHMAN N. Sotos syndrome [J]. Eur J Hum Genet,2007,15(3):264-271.
[10]TATTON-BROWN K,DOUGLAS J,COLEMAN K,et al. Genotype-phenotype associations in Sotos syndrome:an analysis of 266 individuals with NSD1 aberrations [J]. Am J Hum Genet,2005,77(2):193-204.
[11]FICKIE M R,LAPUNZINA P,GENTILE J K,et al.Adults with Sotos syndrome:review of 21 adults with molecularly confirmed NSD1 alterations,including a detailed case report of the oldest person [J]. Am J Med Genet A,2011,155A(9):2105-2111.
[12]KUROTAKI N,HARADA N,SHIMOKAWA O,et al. Fifty microdeletions among 112 cases of Sotos syndrome:low copy repeats possibly mediate the common deletion [J]. Hum Mutat,2003,22(5):378-387.
[13]SANCHEZ R,ZHOU M M. The PHD finger:a versatile epigenome reader [J]. Trends Biochem Sci,2011,36(7):364-372.
[14]QIN S,MIN J. Structure and function of the nucleosome-binding PWWP domain [J]. Trends Biochem Sci,2014,39(11):536-547.
[15]KUDITHIPUDI S,LUNGU C,RATHERT P,et al.Substrate specif i city analysis and novel substrates of the protein lysine methyltransferase NSD1 [J]. Chem Biol,2014,21(2):226-237.
[16]QIAO Q,LI Y,CHEN Z,et al. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation [J]. J Biol Chem,2011,286(10):8361-8368.
[17]RAYASAM G V,WENDLING O,ANGRAND P O,et al. NSD1 is essential for early post-implantation development and has a catalytically active SET domain [J]. EMBO J,2003,22(12):3153-3163.
[18]NAGAI T,MATSUMOTO N,KUROTAKI N,et al. Sotos syndrome and haploinsufficiency of NSD1:clinical features of intragenic mutations and submicroscopic deletions [J]. J Med Genet,2003,(4):285-289.
[19]SAUGIER-VEBER P,BONNET C,AFENJAR A,et al. Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome [J]. Hum Mutat,2007,28(11):1098-1107.
[20]MATSUO T,IHARA K,OCHIAI M,et al.Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome [J]. Am J Med Genet A,2013,161A(1):34-37.
[21]CHOUFANI S,CYTRYNBAUM C,CHUNG B H,et al. NSD1 mutations generate a genome-wide DNA methylation signature [J]. Nat Commun,2015,6:10207.
[22]CHU V T,WEBER T,WEFERS B,et al.Increasing the eff i ciency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells [J]. Nat Biotechnol,2015,33(5):543-548.
