二羟基萘桥联双咪唑盐及其银配合物的合成与结构
2018-05-07赵冬雪赵志翔柳清湘
赵冬雪 ,赵志翔 ,肖 珊 ,柳清湘
(1.天津师范大学 化学学院,天津 300387;2.天津师范大学 无机-有机杂化功能材料化学教育部重点实验室,天津300387;3.天津师范大学 天津市功能分子结构与性能重点实验室,天津 300387)
卡宾又称碳烯,是碳原子上有2个价键连有基团还剩2个未成键电子的高活性中间体.卡宾最早是由Skell于20世纪50年代发现的,1991年,Arduengo等[1]第一次分离得到了稳定的游离N-杂环卡宾(NHC),之后,N-杂环卡宾化学研究得到飞速发展,N-杂环卡宾金属配合物的研究已成为金属有机化学的前沿领域之一[2-7].在N-杂环卡宾金属配合物中,游离状态下寿命短暂的卡宾与金属键合后使之变得稳定,其金属配合物与常用的膦配体金属配合物相比有许多优点,如热、水和空气稳定性高、易制备、结构类型多样化等[8-13].许多N-杂环卡宾金属配合物都具有较高的催化性能,可催化包括C—C偶联反应在内的诸多反应.例如,N-杂环卡宾钯配合物可以作为Suzuki-Miyaura交叉偶联反应、Heck-Mizoroki反应和Sonogashira反应的催化剂,在反应中表现出优良的催化性能[14-16].N-杂环卡宾银或汞配合物在荧光识别领域也备受关注,它们在阳离子客体、阴离子客体和中性客体识别方面都有较好的表现[17-19].此外,一些N-杂环卡宾银、钯、铂的配合物有较高的抗感染和抗癌活性,在生物和药物化学领域应用广泛[20-24].
从近年来对N-杂环卡宾金属配合物的研究可以看出,用以制备N-杂环卡宾金属配合物的配体多选取的是1,3-双取代单咪唑盐[25-27].为了研究咪唑盐及其金属配合物的荧光性能,将强荧光团作为桥联基团引入到咪唑盐中,制备双齿的咪唑盐及其氮杂环卡宾金属配合物,这为N-杂环卡宾金属配合物的研究开拓了新的领域.本研究以荧光团1,5-萘二酚为桥联基团,合成1,5-二[3′-(N-乙基咪唑)丙氧基]萘六氟磷酸盐(Ⅰ),在此基础上,制备其阴离子配合物(Ⅱ)和氮杂环卡宾银配合物(Ⅲ).利用1H NMR、13C NMR和X-线单晶衍射表征了配合物Ⅱ和Ⅲ的结构,并测试了化合物Ⅰ~Ⅲ的荧光发射光谱.
1 实验
1.1 仪器与试剂
仪器:ApexⅡCCD单晶衍射仪,美国Bruker公司;X-4型显微熔点测定仪,北京泰克仪器有限公司;Varian Mercury Vx 400 spectrometer核磁共振仪,美国Varian公司,TMS为内标,DMSO-d6为溶剂.
药品:乙腈、1,3-二溴丙烷、二氯甲烷、N-乙基咪唑、四氢呋喃、六氟磷酸铵、碘化汞、氧化银等,分析纯,天津市光复精细化工有限公司.
1.2 化合物Ⅰ~Ⅲ的合成
化合物Ⅰ~Ⅲ的合成流程如图1所示.在碳酸钾催化下,1,5-萘二酚与1,3-二溴丙烷反应得到1,5-二(3′-溴丙氧基)萘,然后将 1,5-二(3′-溴丙氧基)萘与N-乙基咪唑反应得到1,5-[3′-(N-乙基咪唑)丙氧基]萘二溴化物.以甲醇为溶剂,1,5-[3′-(N-乙基咪唑)丙氧基]萘二溴化物通过与六氟磷酸铵反应得到1,5-二[3′-(N-乙基咪唑)丙氧基]萘六氟磷酸盐(Ⅰ).室温下,化合物Ⅰ分别与碘化汞和氧化银在乙腈中反应,得到阴离子配合物Ⅱ和N-杂环卡宾银配合物Ⅲ.
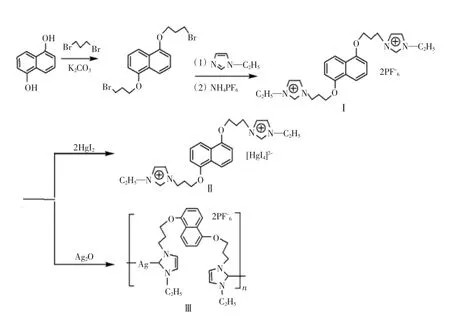
图1 化合物Ⅰ~Ⅲ的合成流程Fig.1 Preparation process of compoundsⅠ-Ⅲ
1.3 1,5-二[3′-(N-乙基咪唑)丙氧基)]萘六氟磷酸盐(Ⅰ)的制备
向 1,5-二羟基萘(1.600 g,10.0 mmol)的乙腈溶液(150mL)中加入碳酸钾(6.900 g,0.05 mol),在搅拌下加热回流2 h,然后加入1,3-二溴丙烷(20.200 g,0.1 mol),在室温下搅拌6 h.将混合物过滤,滤液旋干,在乙醇中结晶,得到浅黄色粉末1,5-二[3′-溴丙氧基]萘.产率:2.400 g(60%).1H NMR(400 MHz,DMSO-d6):δ 2.40 (t,J=6.0,4H,CH2),3.80 (t,J=6.4,4H,CH2),4.26 (t,J=5.6,4H,CH2),7.03 (d,J=7.6,2H,ArH),7.42(t,J=12.8,2H,ArH),7.77(d,J=8.4,2H,ArH).13C NMR(100 MHz,DMSO-d6):δ 65.4(CH2),57.9(CH2),31.8(CH2).
将 N-乙基咪唑(1.150 g,11.9 mmol)和 1,5-二[3′-溴丙氧基]萘(2.000 g,4.9 mmol)加入到 150mL 的四氢呋喃中,在回流下搅拌7 d,过滤产物,用少量四氢呋喃洗涤,得到 1,5-二[3′-(N-乙基咪唑)丙氧基]萘二溴化物的黄色粉末,产率:2.070 g(70%),熔点:154~157℃.
在搅拌下将NH4PF6(1.330 g,8.2 mmol)加入到1,5-二[3′-(N-乙基咪唑)丙氧基]萘二溴化物(2.000 g,3.4 mmol)的甲醇溶液中(100mL),立即形成黄色沉淀,室温下搅拌8 h.过滤收集黄色粉末并用少量甲醇洗涤,得到1,5-二[3′-(N-乙基咪唑)丙氧基]萘六氟磷酸盐(Ⅰ).产率:2.280 g(95%).熔点:166~168℃.1H NMR(400 MHz,DMSO-d6):δ 1.38(t,J=7.2,6H,CH3),2.46(t,J=5.8,4H,CH2),4.17(q,J=7.3,4H,CH2),4.24 (t,J=5.2,4H,CH2),4.48 (q,J=6.6,4H,CH2),7.00(d,J=7.6,2H,ArH),7.42(t,J=8,2H,ArH),7.60 (d,J=8.4,2H,ArH),9.24(s,2H,2-imiH).13CNMR(100MHZ,DMSO-d6):δ153.7(2-imiC),125.1(ArC),124.2(ArC),121.3(ArC),113.9(ArC),65.1(CH2),46.9(CH2),44.1(CH2),29.0(CH2),14.8(CH3).
1.4 阴离子配合物Ⅱ的制备
向化合物Ⅰ(0.200 g,0.3 mmol)的乙腈/DMSO溶液(15mL,V乙腈∶VDMSO=1∶1)中加入碘化汞(0.320 g,0.7 mmol),在室温下反应过夜,将混合物抽滤,所得液体通过旋蒸除去溶剂,残余的浅黄色固体用乙醚洗涤.产率:0.081g(39%).熔点:190~192℃.C26H34HgI4N4O2元素分析理论值:C,27.33%;H,3.00%;N,4.90%.实验值:C,27.47%;H,3.23%;N,4.84%.1HNMR(400MHZ,DMSO-d6):δ 1.38(t,J=7.2,6H,CH3),2.46(t,J=5.8,4H,CH2),4.17(q,J=7.3,4H,CH)2,4.24(t,J=5.2,4H,CH2),4.48(q,J=6.6,4H,CH)2,7.00(d,J=7.6,2H,ArH),7.42(t,J=8.0,2H,ArH),7.60(d,J=8.4,2H,ArH),9.24(s,2H,2-imiH).13C NMR(100 MHZ,DMSO-d6):δ 153.5(2-imiC),65.1(CH2),46.9(CH2),44.1(CH2),29.0(CH2),14.8(CH3).
1.5 配合物Ⅲ的制备
向化合物Ⅰ(0.200 g,0.3 mmol)的乙腈/DMSO 溶液(15mL,V乙腈∶VDMSO=1∶1)中加入氧化银(0.096g,0.4mmol),在室温下避光反应过夜,将混合物抽滤,所得液体通过旋蒸除去溶剂,残余的浅黄色固体用乙醚洗涤.产率:0.078 g(33%).熔点:240~241℃.C26H32AgF6N4O2P元素分析理论值:C,45.56%;H,4.70%;N,8.17%.实验值:C,45.62%;H,4.81%;N,8.33%.1H NMR(400 MHZ,DMSO-d6):δ 1.36 (t,J=13.8,6H,CH3),2.24(s,4H,CH2),4.06(q,J=14.5,8H,CH2),4.26(s,4H,CH2),7.46(s,6H,ArH).13C NMR(100 MHZ,DMSO-d6):δ 153.3(2-imiC),125.5(ArC),124.9(ArC),121.6(ArC),113.5(ArC),64.6(CH2),48.4(CH2),46.0(CH2),30.5(CH2),16.8(CH3).
1.6 X-线单晶衍射
配合物Ⅱ和Ⅲ选定的晶体学数据如表1所示.表1中所有数据均是在Bruker ApexⅡCCD单晶衍射仪上以50 kV和20 mA Mo-Kα射线(λ=0.071 073 nm)测定的,应用SMART和SAINT软件对数据进行采集和整理.在0.6°振荡范围内,θ角的取值范围是1.8°<θ<25°,实证吸收校正采用SADABS程序.采用直接方法解决结构问题,运用SHELXTL和F2方法对所有非氢原子的各向异性进行分析.所有氢原子呈几何结构生长(C—H键键长固定在0.096 nm),分别处于各向异性参数和结构因子的计算中.
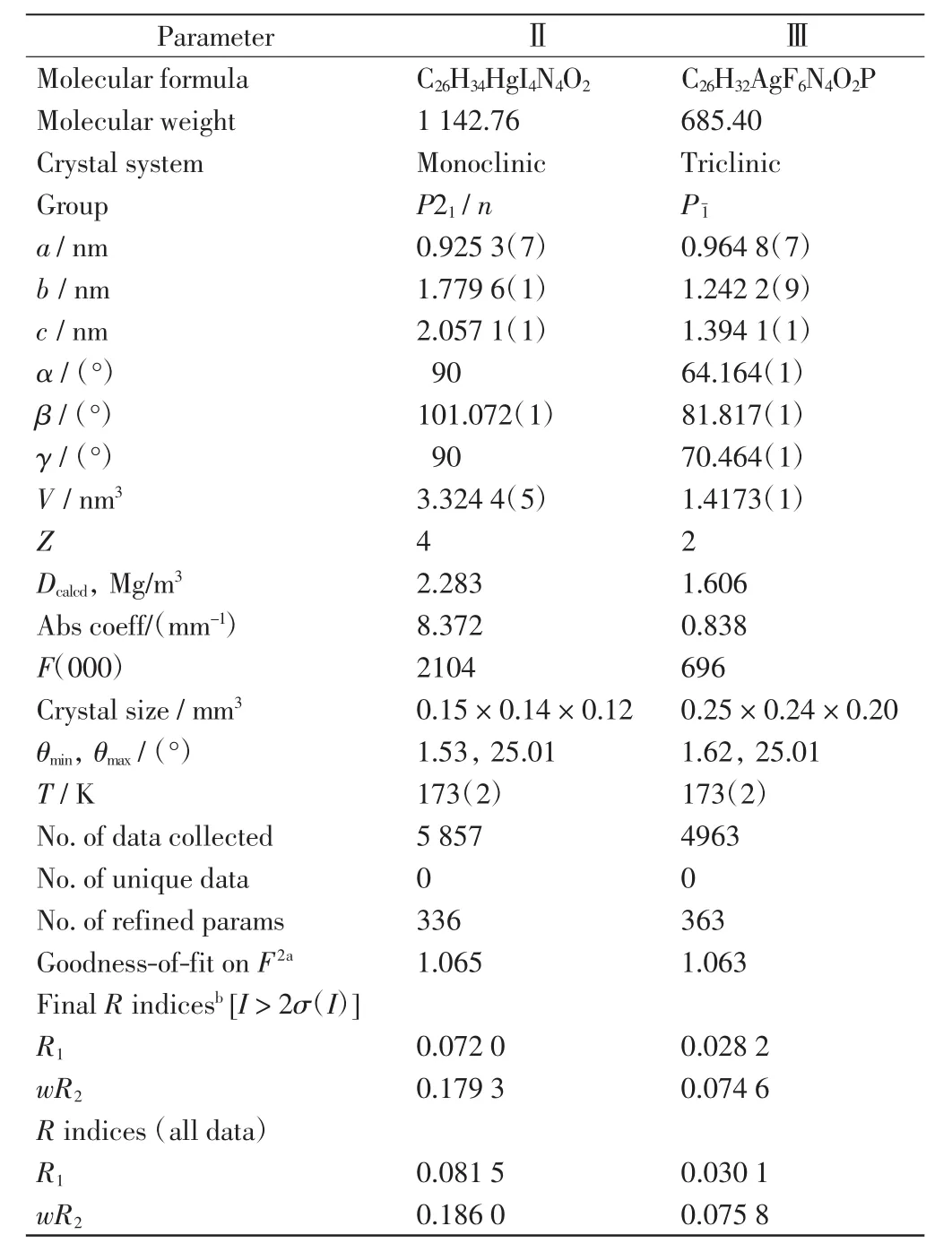
表1 配合物Ⅱ和Ⅲ的晶体数据Tab.1 Summary of crystallographic data for complexesⅡandⅢ
2 结果与分析
2.1 化合物Ⅰ~Ⅲ的表征
化合物Ⅰ对空气和水稳定,溶于常见有机溶剂,几乎不溶于苯、石油醚和水.1H NMR结果显示,化合物Ⅰ咪唑2位上的质子(NCHN)化学位移出现在9.24,与已知的咪唑盐2-位质子化学位移一致[17,28-32].
配合物Ⅱ和Ⅲ对空气与水稳定,溶于二甲基亚砜和乙腈,不溶于乙醚、烃类溶剂和水.乙醚缓慢扩散进入配合物Ⅱ和Ⅲ的乙腈与DMSO溶液中,最终得到符合X-线单晶衍射分析条件的晶体.在配合物Ⅱ的1H NMR分析中,各质子化学位移与化合物Ⅰ的类似.在配合物Ⅲ的1H NMR中,未观测到咪唑2位的质子信号,说明形成了氮杂环卡宾银配合物,其余质子的化学位移与化合物Ⅰ的类似.在配合物Ⅲ的13C NMR中,没有观测到卡宾碳原子的信号,可能是由于卡宾配合物的电子流动性所致[33-34].
2.2 阴离子配合物Ⅱ的结构
阴离子配合物Ⅱ的晶体学结构如图2所示,配合物Ⅱ和Ⅲ的主要键长和键角数据如表2所示,氢键数据如表3所示.
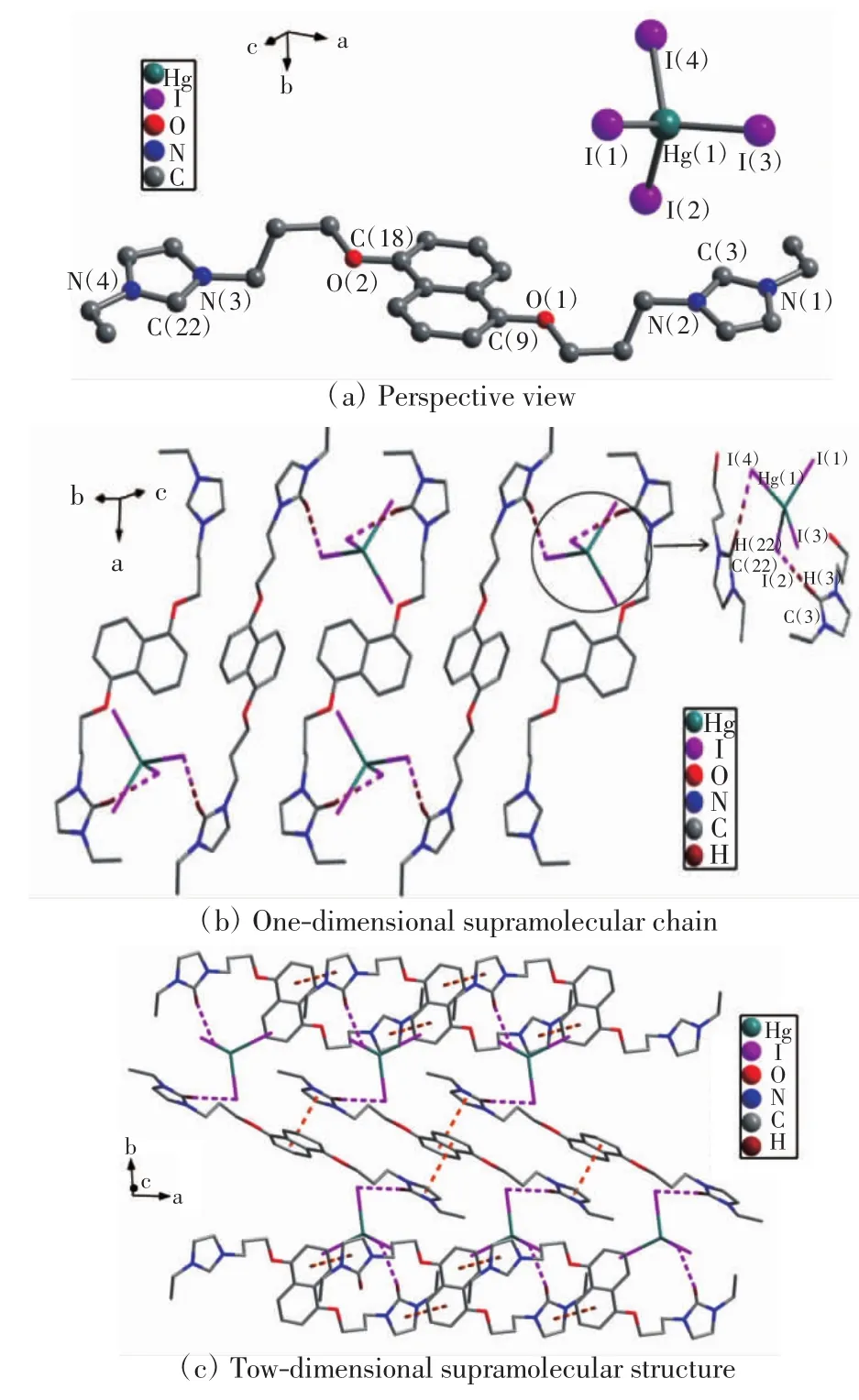
图2 阴离子配合物Ⅱ的晶体结构Fig.2 Crystal structure of anionic complexⅡ
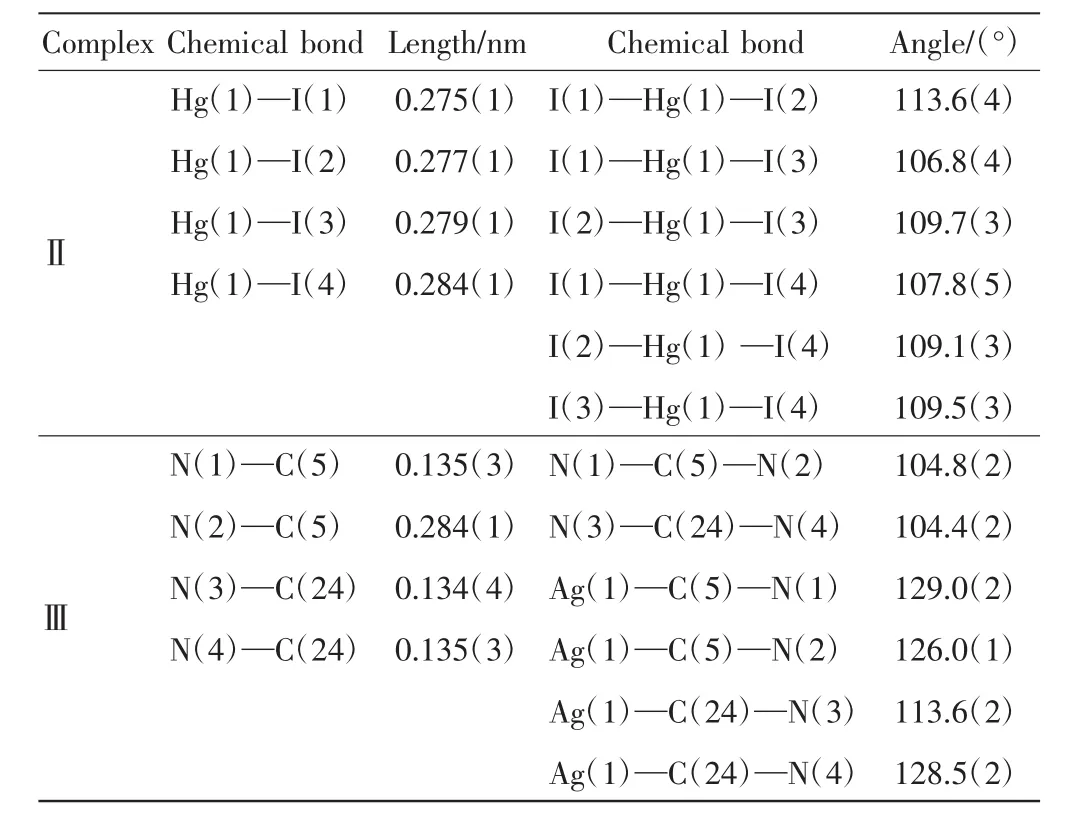
表2 配合物Ⅱ和Ⅲ的部分键长和键角Tab.2 Bond length and bond angle of complexesⅡandⅢ
由图2(a)和表2可以看出,C—N的键长范围为0.130(2)~0.135(2)nm,2个咪唑环的内环角(N—C—N)的键角分别是 108.0(1)°和 108.8(1)°;2 个咪唑环平行并指向相反方向,连接在2个咪唑环上的2个乙基也指向相反方向;萘环和咪唑环形成12.9(6)°的二面角.在阴离子单元[HgI4]2-中,汞离子被4个碘离子包围形成1个四面体构型.4个Hg—I的键长范围为0.275(1)~0.284(1)nm,I—Hg—I键角范围为 107.8(5)°~113(6)°.由图 2(b)可以看出,配合物Ⅱ的一维超分子链是通过C—H…I氢键形成的.在氢键中,氢原子来自咪唑环,碘原子来自[HgI4]2-.由图2(c)可知,一维超分子链通过分子间咪唑环与萘环的π-π堆积作用形成二维超分子结构,π-π堆积作用的面-面距离是0.366(2)nm,中心-中心距离是 0.394(3)nm.

表3 配合物Ⅱ和Ⅲ的氢键几何数据Tab.3 H-Bond geometry for complexesⅡandⅢ
2.3 配合物Ⅲ的结构
配合物Ⅲ的晶体结构如图3所示.配合物Ⅲ的一维链状结构中(图3(a)),C—N的键长范围为0.135(3)~0.135(4)nm,咪唑环内环角(N—C—N)的键角分别是 104.4(2)°和 104.8(2)°,比配合物Ⅱ中的对应值要小.银离子与卡宾碳相连,C—Ag键长为0.208(3)nm,C—Ag—C 近似线型排布,键角为 177.6(1)°.同一个配体中的2个咪唑环的二面角是50.9(1)°,萘环和2 个咪唑环的二面角分别为 2.6(1)°和47.5(2)°,同一个NHC—Ag—NHC单元中2个咪唑环的二面角是50.9(1)°.所有萘环平行,在银原子同一侧的所有咪唑环也是平行的.相邻Ag…Ag的距离是0.964(8)nm.由图3(b)可知,配合物Ⅲ的二维超分子层通过C—H…F氢键形成.在氢键中,氢原子来自配合物的咪唑环,氟原子来自.另外,二维超分子层通过分子间咪唑环与萘环的π-π堆积作用进一步扩展成三维超分子框架(图3(c)),咪唑环与萘环的面-面距离是0.352(3)nm,中心-中心距离是 0.359(2)nm.
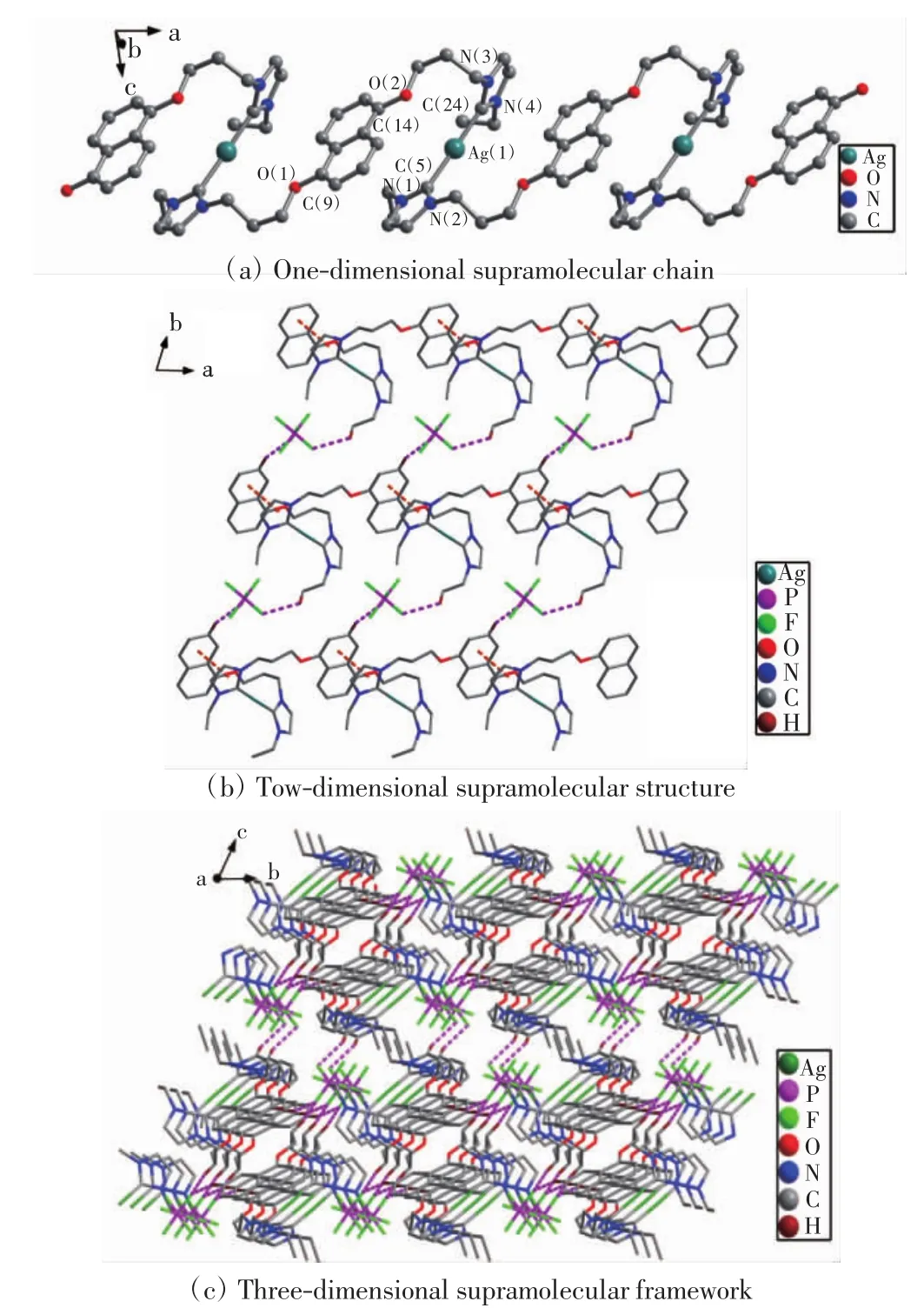
图3 配合物Ⅲ的晶体结构Fig.3 Crystal structure of complexⅢ
2.4 化合物Ⅰ~Ⅲ的荧光特征
在激发波长为232 nm、狭缝宽度为3 nm的条件下测试化合物Ⅰ~Ⅲ在乙腈溶液中的荧光发射光谱(浓度为1×10-6mol/L),结果如图4所示.

图4 化合物Ⅰ~Ⅲ的荧光发射光谱图Fig.4 Fluorescence emission spectra of compoundsⅠ-Ⅲ
由图4可以看出,化合物Ⅰ~Ⅲ在320~385 nm的波长范围内具有相似的荧光发射峰,并且在342 nm左右均达到最大发射强度.3种化合物中,配合物Ⅱ的荧光强度略高于化合物Ⅰ,表明阴离子的改变对Ⅰ的荧光发射光谱并没有明显影响,即汞离子的加入并未导致配合物Ⅱ中重离子效应的出现.与化合物Ⅰ相比,配合物Ⅲ的荧光强度有较大幅度的提高,表明氮杂环卡宾银配合物的生成对化合物Ⅰ的荧光发射光谱有较大影响.
3 结论
本研究合成了萘基桥联的双咪唑盐Ⅰ及其阴离子配合物Ⅱ和氮杂环卡宾银配合物Ⅲ,对配合物Ⅱ和Ⅲ的晶体结构进行了表征.在配合物Ⅱ的分子结构中,2个咪唑环是平行的,萘环和这2个咪唑环形成12.9(6)°的二面角.在配合物Ⅲ的一维聚合链中,所有萘环是平行的,在银原子同一侧的所有咪唑环也是平行的,相邻Ag…Ag的距离是0.964(8)nm.在配合物Ⅱ和Ⅲ的晶体堆积中,它们的二维超分子层和三维超分子建筑是通过分子间弱相互作用形成的.此外,化合物Ⅰ~Ⅲ的荧光发射光谱表明,氮杂环卡宾银配合物Ⅲ的荧光强度比咪唑盐I的荧光强度有了显著增强.
参考文献:
[1]ARDUENGO ⅢIII A J,HARLOW R L,KLINE M K.A stable crystalline carbene[J].J Am Chem Soc,1991,113(1):361-363.
[2]COLE M L,JONES C,JUNK P C.Studies of the reactivity of N-heterocyclic carbenes with halogen and halide sources[J].New J Chem,2002,26(10):1296-1303.
[3]HERRMANN W A,KÖCHER C,GOOSSEN L,et al.Heterocyclic carbenes:Ahigh-yieldingsynthesisofnovel,functionalizedN-heterocyclic carbenes in liquid ammonia[J].Chem Eur J,1996,2(12):1627-1636.
[4]NYCE G W,CSIHONY S,WAYMOUTH R M,et al.A general and versatile approach to thermally generated N-heterocyclic carbenes[J].Chem Eur J,2004,10(16):4073-4079.
[5]OTTO M,CONEJERO S,CANAC Y,et al.Mono-and diaminocarbenes from chloroiminium and-amidinium salts:Synthesis of metal-free bis(dimethylamino)carbene[J].J Am Chem Soc,2004,126(4):1016-1017.
[6]SCOTT N M,NOLAN S P.Stabilization of organometallic species achieved by the use of N-heterocyclic carbene(NHC)ligands[J].Eur J Inorg Chem,2005,2005(10):1815-1828.
[7]CRUDDEN C M,ALLEN D P.Stability and reactivity of N-heterocyclic carbenecomplexes[J].CoordChemRev,2004,248(21/22/23/24):2247-2273.
[8]MCGUINNESS D S,CAVELL K J.Donor-functionalized heterocyclic carbene complexes of palladium(Ⅱ):Efficient catalysts for C—C coupling reactions[J].Organometallics,2000,19(19):741-748.
[9]WANG X,LIU S,JIN G X.Preparation,structure,and olefin polyme-rization behavior of functionalized nickel(Ⅱ)N-heterocyclic carbene complexes[J].Organometallics,2004,23(25):6002-6007.
[10]ROSEN E L,SUNG D H,CHEN Z,et al.Olefin metathesis catalysts containing acyclic diaminocarbenes[J].Organometallics,2014,29(1):250-256.
[11]SAMOJLOWICZ C,BIENIEK M,GRELA K.Ruthenium-based olefin metathesis catalysts bearing N-heterocyclic carbene ligands[J].Chem Rev,2009,109(9):3708-3742.
[12]PERIS E,LOCH J A,MATA J,et al.A Pd complex of a tridentate pincer CNC bis-carbene ligand as a robust homogenous Heck catalyst[J].Chem Commun,2001,32(20):201-202.
[13]MCGUINNESS D S,GIBSON V C,STEED J W.Bis(carbene)pyridine complexes of the early to middle transition metals:Survey of ethylene oligomerizationandpolymerizationcapability[J].Organometallics,2004,23(26):6288-6292.
[14]EZUGWU C I,MOUSAVI B,ASRAF M A,et al.Post-synthetic modified MOF for Sonogashira cross-coupling and knoevenagel condensation reactions[J].J Catalysis,2016,344:445-454.
[15]LIU Q X,ZHANG W,ZHAO X J,et al.NHC PdII complex bearing 1,6-hexylenelinker:SynthesisandcatalyticactivityintheSuzuki-Miyaura and Heck-Mizoroki reactions[J].Eur J Org Chem,2013,44(35):1253-1261.
[16]LIU Q X,ZHAO L X,ZHAO X J,et al.Silver(I),palladium(Ⅱ)and mercury(Ⅱ)NHC complexes based on bis-benzimidazole salts with mesitylene linker:Synthesis,structural studies and catalytic activity[J].J Organomet Chem,2013,731(731):35-48.
[17]LIU Q X,WEI Q,LIU R,et al.NHC macrometallocycles of mercury(Ⅱ)and silver(Ⅰ):Synthesis,structural studies and recognition of Hg(Ⅱ)complex4forsilverion[J].RSCAdv,2015,5(36):28435-28447.
[18]LIU Q X,CHEN J R,SUN X F,et al.An NHC silver(Ⅰ)macrometallocycle:Synthesis,structure and selective recognition of iodide anions[J].RSC Adv,2016,6(15):12256-12262.
[19]LIU Q X,SUN X F,ZHAO D X,et al.Selective recognition of m-dinitrobenzene based on NHC silver(Ⅰ)macrometallocycle[J].Sensors and Actuators B,2017,249:203-209.
[20]HINDI K M,PANZNER M J,TESSIER C A,et al.The medicinal applicationsofimidazoliumcarbene-metalcomplexes[J].ChemRev,2009,109(8):3859-3884.
[21]KASCATAN-NEBIOGLU A,PANZNER M J,TESSIER C A,et al.N-heterocyclic carbene-silver complexes:A new class of antibiotics[J].Coord Chem Rev,2007,251(5/6):884-895.
[22]OEHNINGER L,RUBBIANI R,OTT I.N-heterocyclic carbene metal complexesinmedicinalchemistry[J].DaltonTrans,2013,42(10):3269-3284.
[23]PATILSA,PATILS A,RANGAPPA R P,et al.N-heterocyclic carbene metal complexes as bio-organometallic antimicrobial and anticancer drugs[J].Future Med Chem,2015,7(10):1305-1333.
[24]KAPDI A R,FAIRLAMB I J S.Anti-cancer palladium complexes:A focusonPdX2L2,palladacyclesandrelated complexes[J].ChemSocRev,2014,43(13):4751-4777.
[25]MARCEL B,FRANK R,PETER H.1,2-Bis(di-tert-butylphosphino)imidazole(dtbpi):Aversatileimidazole-based,rigid,bulkybisphosphine ligandfortransitionmetals[J].Organometallics,2015,34(2):506-521.
[26]VEDHAGIRI K,VIVEK G,GANAPATHI A.Synthesis,structure,and coordination chemistry of phosphine functionalized imidazole/imidazolium salts and cleavage of a C—P bond in an NHC-phosphenium salt using a Pd(0)precursor[J].Organometallics,2015,34(15):3713-3720.
[27]VEDHAGIRI K,VIVEK G,GANAPATHI A.Synthesis of imidazolebased functionalized mesoionic carbene complexes of palladium:Comparison of donor properties and catalytic activity toward Suzuki-Miyaura coupling[J].Organometallics,2014,33(21):6218-6222.
[28]MCGUINNESS D S,MUELLER W,WASSERSCHEID P,et al.Nickel(Ⅱ)heterocyclic carbene complexes as catalysts for olefin dimerization in an imidazolium chloroaluminate ionic liquid[J].Organometallics,2002,21(1):175-181.
[29]KURDZIEL K,GLOWIAK T.X-ray and spectroscopic characterization ofoctahedralcobalt(Ⅱ)andnickel(Ⅱ)complexeswith1-allylimidazole in the solid state and electron-donor properties of the latter in aqueous solution[J].Polyhedron,2000,19(20/21):2183-2188.
[30]LIU Q X,YAO Z Q,ZHAO X J,et al.NHC metal (silver,mercury,and nickel)complexes based on quinoxaline-dibenzimidazolium salts:Synthesis,structural studies,and fluorescent chemosensors for Cu2+by charge transfer[J].Organometallics,2013,32(12):3493-3501.
[31]LIU Q X,LIU R,DING Y,et al.Preparation and structure of NHC Hg(Ⅱ)andAg(Ⅰ)macrometallocycles[J].CrystEngComm,2015,17(48):9380-9393.
[32]LIU Q X,CAI K Q,ZHAO Z X,et al.Synthesis,structure and catalysis of a NHC-Pd (II)complex based on a tetradentate mixed ligand[J].RSC Adv,2015,5(104):85568-85578.
[33]LEE C K,LEE K M,LIN I J B.Inorganic-organic hybrid lamella of diand tetra-nuclear silver-carbene complexes[J].Organometallics,2002,21(21):10-12.
[34]GARRISON J C,SIMONS R S,TALLEY J M,et al.Synthesis and structural characterization of an imidazolium-linked cyclophane and the silver complex of an N-heterocyclic carbene-linked cyclophane[J].Organometallics,2001,20(7):1276-1278.
