缺氧诱导因子与糖尿病肾病
2018-05-05杜小红综述谢红浪审校
杜小红 综述 谢红浪 审校
缺氧诱导因子(HIF)是机体适应缺氧的关键转录因子,其对糖类等物质的调节在一定程度上可改善机体能量代谢,保证低氧下的细胞代谢平衡。大量研究表明[1-2],除低氧之外,高糖可能是调控HIF表达的另一重要因子,糖尿病肾病(DN)时HIF表达明显增加,能进一步加重肾脏纤维化、促进终末期肾病发生。而二甲双胍抑制HIF表达作用,将成为其治疗DN的重要机制之一。本文拟就低氧下的糖代谢变化,高糖及二甲双胍对HIF的调节及其作用机制进行综述,为DN的治疗新靶点提供理论依据。
HIF-1的结构及生物学特点
HIF是一种异源蛋白二聚体,由HIF-α和HIF-β两个亚基组成。其中,α亚基的活性受缺氧信号的调控,β亚基在细胞内稳定表达;只有α活性亚基与β亚基相互连接形成异二聚体时,HIF才可转移到细胞核内与缺氧反应元件(HRE)的DNA序列相结合,调节多个目标基因的转录。在正常氧供时,依赖机体内氧及铁的HIF脯氨酰羟化(PHD)酶可诱导HIF-α亚基上氧依赖区域(ODD)内的脯氨酸残基羟基化,羟基化的HIF继而被希佩尔-林道(von Hippel-Lindau,VHL)泛素连接酶辨识并泛素化,最后被蛋白酶体快速降解。因此,正常情况下细胞内基本检测不出α亚基的存在;在低氧状态下,HIF-α亚基才可以避免泛素蛋白酶体系统的降解作用,维持HIF-1的转录活性。而具有转录活性的核蛋白HIF-1能调节参与细胞内物质代谢的重要因子转录,适应低氧环境[3]。
HIF对糖代谢的影响
长期低氧适应能影响糖代谢,如糖摄取、利用、氧化等作用增强,虽其具体作用机制尚不明确,但目前认为HIF主要通过上调糖代谢相关基因表达,调节糖代谢过程。
增强葡萄糖转运体蛋白(GLUT)表达缺氧情况下细胞主要依赖葡萄糖无氧糖酵解供能,其中葡萄糖通过载体跨膜转入胞内是细胞糖代谢的限速步骤;而GLUT1作为葡萄糖跨膜转运的主要载体,其在细胞膜上表达量可能是机体适应缺氧、缺血等应激状态的一个重要调控机制。早期即发现GLUT1基因片段中含HIF结合位点(HBS),缺氧可诱导GLUT1 mRNA表达,上调GLUT1水平。Ouiddir等[4]证实低氧能诱导肺泡上皮细胞的GLUT1活性增加;且GLUT1 mRNA的转录表达呈缺氧浓度、时间依赖性,即基因表达随氧浓度降低和缺氧时间延长而显著增加。而Fujino等[5]同样证明HIF促进GLUT1的表达。
除了直接诱导GLUT表达,HIF介导的胰岛素及胰岛素受体表达和敏感性增强,间接促进GLUT转位和转运的能力增强。由于胰岛素与其受体的结合,可以促进囊泡内GLUT发生细胞膜转位效应,提高膜上的葡萄糖转运体密度。Chen等[6]研究发现,与久居高海拔人群的血清胰岛素水平显著升高不同,暴露于高海波(约5 000米)的大鼠体内胰岛素水平虽降低,胰高血糖素显著升高,但胰岛素敏感性的增加仍然导致血糖水平的显著下降。因此胰岛素、胰岛素受体数量以及胰岛素敏感性均影响GLUT转位,从而影响葡萄糖代谢。GLUT的表达、储存、转位增加是低氧下HIF调节葡萄糖摄取的基础。
增加糖酵解实验证实,为弥补低氧时的产能不足,机体内聚集的HIF通过低氧诱导反应下调有氧磷酸化过程,调节细胞糖酵解过程中关键酶的表达[7],如已糖激酶(HK)、乳酸脱氢酶A(LDHA)、丙酮酸脱氢酶激酶1(PDK 1)等,增加糖酵解活性。但脯氨酰羟化酶3(PHD3)介导的丙酮酸激酶M2(PKM2)羟基化可促进糖酵解和HIF间的相互作用,认为这是HIF在缺氧条件下发挥功能的首要条件;Luo等[8]发现定位在细胞核上的PKM2可与HIF直接结合并相互提高转录水平,形成正反馈调节。与正常细胞不同,肿瘤细胞在体内主要依赖糖酵解进行代谢,产生乳酸;称之为Warburg效应。而PKM2在增殖细胞中高度表达,尤其是肿瘤细胞,这对肿瘤细胞代谢极其重要。除此之外,肿瘤患者体内突变或过度表达的癌基因Myc与HIF的相互作用能提高糖酵解相关基因的转录活性,促进葡萄糖摄取[9]。由此可知,低氧时活化的HIF能够增加糖酵解活性。
DN时HIF的变化
在小鼠DN模型的肾组织中,实验者利用4′,6-二脒基-2-苯基吲哚(DAPI)进行细胞核染色,并通过免疫组化法检测HIF表达,结果发现DN肾组织中HIF水平较正常组明显上升[1-2]。并在链脲霉素诱导的1型糖尿病和db/db型的2型糖尿病小鼠模型中[1],及高糖处理的海马神经元内[10],均表现出HIF的上调。故高糖是除低氧外,影响DN时HIF表达的另一重要因素。关于DN时HIF的改变,主要通过以下两种信号通路进行调控。
ChRE/HIF mRNA通路Isoe等[1]发现糖尿病小鼠模型中肾小球系膜细胞内HIF表达增加;且DN时HIF的表达呈血糖依耐性上升;而同等渗透压下的非糖物质组内HIF表达并无明显改变,故认为HIF变化与高血糖有关,且主要与HIF-1αmRNA的转录增加相关。由于人类及小鼠的HIF-1α基因序列含有碳水化合物反应元件(ChRE),即葡萄糖作用位点[11];实验发现ChRE能介导高糖条件下HIF-1αmRNA的表达上调[1]。这可能是因为碳水化合物反应元件结合蛋白(ChREBP)作为一类与ChRE结合的转录因子,可以调节细胞及组织内葡萄糖代谢;而高糖通过刺激胞内ChREBP转入核内,参与系膜细胞内HIF及其目标基因的表达调控[12]。目前认为高糖通过促进HIF-1αmRNA转录上调HIF水平,这与低氧防止HIF降解,保证HIF含量稳定的作用机制不同。
AGE/Morg1/PHD3/HIF通路晚期糖基化终末产物(AGE)与其受体(RAGE)结合抑制丝裂原活化蛋白激酶(MAPK)组织蛋白1(Morg1)表达是DN患者体内HIF水平上调的另一机制。DN时,AGEs可进一步促进足细胞内血管紧张素Ⅱ(AngⅡ)表达,AngⅡ作为G蛋白偶联受体激动剂,通过激活MAPK信号通路,影响MAPK酶联反应中支架蛋白Morg1的表达。而Morg1作为PHD3激活剂,能抑制HIF表达。Bondeva等[2]证实与对照组相比,AGE-BSA作用下的Morg1 mRNA表达显著抑制,且主要集中于RAGE高表达的小鼠近端肾小管细胞(MTC)及足细胞内,表明AGE显著抑制Morg1表达;同时HZ(Morg1+/-)小鼠体内的HIF表达显著高于WT(Morg1+/+)小鼠。因此,DN时,高表达的AngⅡ与AGE/RAGE,通过激活Morg1/PHD3/HIF信号通路,上调体内HIF表达。
此外,肾小球高压、旁分泌或内分泌因素以及氧化应激等也影响DN时HIF的水平。
而基因多态性作为HIF-1α的重要生物特性,与多种疾病的发生发展密切相关,如肿瘤、糖尿病、心血管疾病等。但近期研究证实,HIF-1α的Pro582Ser多态性(rs11549465)与DN密切相关[13-14]。这主要是由于HIF-1α基因外显子存在rs11549465多态性,其中C→T突变诱导脯氨酸变为丝氨酸(Pro582Ser);在缺氧、高糖两者因素的共同作用下,CT及TT基因型的转录水平将显著高于CC基因型,最终影响HIF-1α的表达及其对下游信号基因的调控。此外,Bi等[14]指出HIF-1α的基因多态性对DN不同阶段影响存在差异性。DN早期,上调的HIF-1α通过促进血红素加氧酶1、糖酵解酶等靶基因的表达,诱导机体适应低氧状态,发挥肾保护作用;当DN发展到一定阶段,过度表达的HIF-1α反而加重肾纤维化程度。这可能与以下机制相关[15]:首先,活化的HIF能刺激炎症细胞的增殖,并集聚于肾损部位,这对肾脏成纤维瘢痕的形成至关重要;其次,HIF可与促纤维化的下游信号基因相结合,如胶原蛋白1、纤溶酶原激活物抑制剂1(PAI1)、结缔组织生长因子(CTGF)等,导致间质胶原的生成,减少细胞外基质(ECM)的降解。除ECM的沉积作用外,HIF还参与肾小管上皮间质转化(EMT)过程,它通过促进间质信号表达,抑制上皮信号传导,继而诱导ECM的合成[16]。而逐渐积聚的ECM最终将破坏正常肾组织,促进肾纤维化。
二甲双胍对HIF的影响
Nayak等[17]发现抑制HIF活性可减轻DN的蛋白尿及肾纤维化程度,故干预HIF可能成为延缓DN进展的重要机制。最新研究指出,二甲双胍能减轻DN中蛋白尿的肾毒性损害,产生肾保护作用[18-19]。作为2型糖尿病的一线治疗药物,其主要作用是抑制肝糖原的产生[20]。但近年来,二甲双胍在癌症治疗领域的应用受到了广泛关注[21-22]。在糖尿病的降糖治疗中,研究者发现二甲双胍能显著降低肿瘤风险,改善患者生存率[21],这可能与HIF机制相关。由于缺氧作为肿瘤生存微环境的主要特征,能显著影响其临床治疗效果[23]。Li等[24]通过实验证实,二甲双胍有效抑制低氧环境下HIF表达。除直接解除高糖导致HIF 表达增加,二甲双胍还通过以下机制间接调控低氧下HIF水平。
降低氧耗减少HIF表达二甲双胍是线粒体呼吸链中复合物Ⅰ的抑制剂,在小剂量时即可抑制其活性。为模拟二甲双胍对HIF的调控,Takiyama等[25]分别利用线粒体丙酮酸转运体抑制剂(CHC)、mTOR抑制剂(雷帕霉素)、线粒体呼吸链酶复合体1、3抑制剂(鱼藤酮、抗霉素A)分别对小鼠细胞进行预处理,发现CHC及雷帕霉素并未影响HIF的水平,而呼吸链酶抑制剂能显著抑制HIF表达。且Zhao等[26]对小鼠进行低氧敏感染料(哌莫硝唑),染色处理,结果显示口服二甲双胍实验鼠体内低氧染色较对照组明显减弱,进一步证实二甲双胍通过抑制线粒体氧耗,减少ATP生成,提高细胞氧合作用,抑制HIF表达。
抑制HIF聚集及转录表达二甲双胍能抑制低氧下HIF聚集。Zhou等[27]发现二甲双胍对HIF的抑制作用呈剂量-时间依赖性。与低氧下HIF表达增加相比,二甲双胍能降低肝细胞内HIF水平,且保持持续抑制状态;而当二甲双胍浓度超过10 mmol/L时,HIF表达将完全被抑制。除此之外,二甲双胍抑制HIF相关转录蛋白的表达,如GLUT1、VEGF等。与既往结论不同,该实验显示二甲双胍处理组中HIF-mRNA的水平并无改变。故证实二甲双胍诱导的HIF减少受翻译后机制调控。
早期有研究发现,AMPK-mTOR-HIF信号通路是二甲双胍抑制肿瘤生长的主要机制[28-29]。Takiyama等[25]证实缺氧下的AMPK激活剂(AICAR)虽能增加AMPK磷酸化,减少磷酸化的mTOR,但并不抑制HIF表达。Zhou等[27]利用小干扰RNA(siRNA)及AMPK抑制剂(复合物 C)诱导AMPK的失活,结果显示与单用二甲双胍治疗组相比,实验组HIF水平并未显著增加,即失活的AMPK并没有解除二甲双胍对HIF的抑制作用。表明二甲双胍对HIF的调控并非依赖AMPK的调控。由于HIF水平受合成及降解两方面的调节,故研究者利用蛋白酶抑制剂MG-132进行干预,发现MG-132能够解除二甲双胍对HIF的抑制作用,HIF水平较前升高,再次证实二甲双胍通过诱导蛋白酶体对HIF-1α的降解来抑制HIF聚集(图1)[25]。
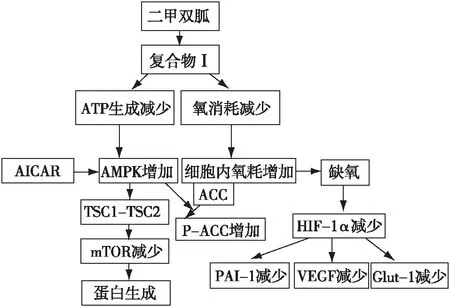
图1 二甲双胍调节HIF-1水平[26]二甲双胍通过抑制线粒体呼吸抑制低氧诱导的HIF-1α蛋白质表达。二甲双胍通过抑制线粒体氧耗导致细胞内的氧气再分配,促进HIF-1α蛋白降解;ATP消耗减少能激活AMPK,诱导AMPK-mTOR信号通路的激活,促进相关蛋白的合成。因此,AICAR本身并不抑制低氧诱导的HIF-1α表达;HIF:缺氧诱导因子;AMPK:腺苷酸活化蛋白激酶;ATP:三磷酸腺苷;AICAR:AMPK依赖性蛋白激酶激动剂;TSC:结节性硬化复合物;mTOR:哺乳动物雷帕霉素靶蛋白;ACC:抗乙酰辅酶A羟化酶;PAI-1:纤溶酶原激活物抑制剂1;VEGF:血管内皮生长因子;Glut-1:葡萄糖转运体1
尽管二甲双胍对HIF的抑制导致低氧下GLUT1表达减少,影响体内糖代谢水平;但二甲双胍诱导的AMPK活化可以提高GLUT1/4及糖酵解相关酶的水平,促进细胞或组织对葡萄糖的利用[30],同时解除高血糖本身通过解偶联途径增加氧耗,改善机体缺氧状态。除AMPK作用外,二甲双胍对线粒体呼吸抑制能导致细胞氧化磷酸化的减弱,诱导巴斯德反应,产生低氧适应。肾缺氧作为DN的早期表现,亦是所有肾脏疾病的共同代谢途径。因此,在慢性肾脏病,尤其是DN中,二甲双胍显著改善肾近端小管上皮细胞内缺氧及氧化应激状态,抑制HIF表达,产生肾保护作用。
小结:HIF是一类具有转录活性的核蛋白,调节体内糖代谢过程。高糖水平是影响HIF及其目标基因转录的另一重要因素。缺氧和肾小管间质纤维化是慢性肾脏疾病的共同病理特点,受活化HIF的调控。与其他肾脏疾病相比,DN时HIF增加更明显,敲除HIF可能成为DN新的治疗手段。二甲双胍作为一线降糖药,除能解除高糖对HIF的诱导作用,亦通过减少氧耗、促进蛋白酶解,降低DN患者体内的HIF水平,从而有可能延缓或阻止肾纤维化,改善肾脏损害进展。同时HIF在血管生物学方面的核心作用,也为DN患者心血管并发症的预防及诊治提供新靶点。
1 Isoe T,Makino Y,Mizumoto K,et al.High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein.Kidney Int,2010,78(1):48-59.
2 Bondeva T,Heinzig J,Ruhe C,et al.Advanced glycated end-products affect HIF-transcriptional activity in renal cells.Mol Endocrinol,2013,27(11):1918-1933.
3 Lefere S,Van Steenkiste C,Verhelst X,et al.Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease.Cell Mol Life Sci,2016,73(18):3419-3431.
4 Ouiddir A,Planès C,Fernandes I,et al.Hypoxia upregulates activity and expression of the glucose transporter GLUT1 in alveolar epithelial cells.Am J Respir Cell Mol Biol,1999,21(6):710-718.
5 Fujino M,Aishima S,Shindo K,et al.Expression of glucose transporter-1 is correlated with hypoxia-inducible factor 1α and malignant potential in pancreatic neuroendocrine tumors.Oncol Lett,2016,12(5):3337-3343.
6 Chen XQ,Dong J,Niu CY,et al.Effects of hypoxia on glucose,insulin,glucagon,and modulation by corticotropin-releasing factor receptor type 1 in the rat.Endocrinology,2007,148(7):3271-3278.
7 Semenza GL.HIF-1: upstream and downstream of cancer metabolism.Curr Opin Genet Dev,2010,20(1):51-56.
8 Luo W,Hu H,Chang R,et al.Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1.Cell,2011,145(5):732-744.
9 Semenza GL.Regulation of metabolism by hypoxia-inducible factor 1.Cold Spring Harb Symp Quant Biol,2011,76:347-353.
10 Lee HJ,Ryu JM,Jung YH,et al.High glucose upregulates BACE1-mediated Aβ production through ROS-dependent HIF-1α and LXRα/ABCA1-regulated lipid raft reorganization in SK-N-MC cells.Sci Rep,2016,6:36746.
11 Uyeda K,Repa JJ.Carbohydrate response element binding protein,ChREBP,a transcription factor coupling hepatic glucose utilization and lipid synthesis.Cell Metab,2006,4(2):107-110.
12 Chang M L,Chiu C J,Shang F,et al.High glucose activates ChREBP-mediated HIF-1α and VEGF expression in human RPE cells under normoxia//Retinal Degenerative Diseases.Springer New York,2014: 609-621.
13 Gu HF,Zheng X,Abu SN,et al.Impact of the hypoxia-inducible factor-1 α (HIF1A) Pro582Ser polymorphism on diabetes nephropathy.Diabetes Care,2013,36(2):415-421.
14 Bi YX,Yu L,Jin GX.Correlation between polymorphisms of hypoxia-inducible factor-1α Pro582Ser and type 2 diabetic nephropathy.Genet Mol Res,2015,14(4):14503-14509.
15 Liu M,Ning X,Li R,et al.Signalling pathways involved in hypoxia-induced renal fibrosis.J Cell Mol Med,2017,21(7):1248-1259.
16 Grande MT,Sánchez-Laorden B,López-Blau C,et al.Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease.Nat Med,2015,21(9):989-997.
17 Nayak BK,Shanmugasundaram K,Friedrichs WE,et al.HIF-1 Mediates Renal Fibrosis in OVE26 Type 1 Diabetic Mice.Diabetes,2016,65(5):1387-1397.
18 Ravindran S,Kuruvilla V,Wilbur K,et al.Nephroprotective Effects of Metformin in Diabetic Nephropathy.J Cell Physiol,2017,232(4):731-742.
19 Allouch S,Munusamy S.Metformin attenuates albumin-induced alterations in renal tubular cells in vitro.J Cell Physiol,2017,232(12):3652-3663.
20 Foretz M,Guigas B,Bertrand L,et al.Metformin: from mechanisms of action to therapies.Cell Metab,2014,20(6):953-966.
21 Rêgo DF,Pavan LM,Elias ST,et al.Effects of metformin on head and neck cancer: a systematic review.Oral Oncol,2015,51(5):416-422.
22 Harada K,Ferdous T,Harada T,et al.Metformin in combination with 5-fluorouracil suppresses tumor growth by inhibiting the Warburg effect in human oral squamous cell carcinoma.Int J Oncol,2016,49(1):276-284.
23 Wigerup C,Påhlman S,Bexell D.Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer.Pharmacol Ther,2016,164:152-169.
24 Li X,Li J,Wang L,et al.The role of metformin and resveratrol in the prevention of hypoxia-inducible factor 1α accumulation and fibrosis in hypoxic adipose tissue.Br J Pharmacol,2016,173(12):2001-2015.
25 Takiyama Y,Harumi T,Watanabe J,et al.Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1α expression and oxygen metabolism.Diabetes,2011,60(3):981-992.
26 Zhao W,Li A,Feng X,et al.Metformin and resveratrol ameliorate muscle insulin resistance through preventing lipolysis and inflammation in hypoxic adipose tissue.Cell Signal,2016,28(9):1401-1411.
27 Zhou X,Chen J,Yi G,et al.Metformin suppresses hypoxia-induced stabilization of HIF-1α through reprogramming of oxygen metabolism in hepatocellular carcinoma.Oncotarget,2016,7(1):873-884.
28 Li W,Saud SM,Young MR,et al.Targeting AMPK for cancer prevention and treatment.Oncotarget,2015,6(10):7365-7378.
29 Han G,Gong H,Wang Y,et al.AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell.Cancer Biol Ther,2015,16(1):77-87.
30 Fujii N,Jessen N,Goodyear LJ.AMP-activated protein kinase and the regulation of glucose transport.Am J Physiol Endocrinol Metab,2006,91(5):E867-E877.
