伊曲康唑无定型固体分散体的制备及体外评价
2018-05-04洪子越施沈一郭玉申刘建平
洪子越,施沈一,郭玉申,刘建平*
(1中国药科大学药剂系,南京210009;2上海合全药业股份有限公司,上海200131)
伊曲康唑(itraconazole,ITZ)作为广谱三唑类抗真菌药,是目前临床上治疗真菌感染的常用药物。它可与真菌细胞色素P450发生交互作用,阻断去甲基化过程,抑制真菌细胞膜必需成分麦角固醇的生物合成,使真菌生长受到抑制,从而发挥其抗真菌作用[1]。伊曲康唑属于典型的生物药剂学分类系统(BCS)Ⅱ类药物,在pH 1.2的模拟胃液(SGF)、去离子水和pH 6.8的模拟肠液(SIF)中的溶解度分别是3.9,0.002和0.003μg/mL。据报道,由于其溶解度差,过饱和ITZ体系在溶出过程中快速发生沉淀,导致生物利用度降低[2]。因此,提高ITZ的溶解度和体外溶出度非常重要。
提高溶解度的手段主要包括成盐、多晶、前药、微粉化、制备固体分散体和包合物等[3]。其中固体分散体(solid dispersion,SD)技术是提高难溶性药物溶解度、溶出度和生物利用度的有效手段之一[4]。通过将一种或多种药物活性成分(active pharmaceutical ingredients,APIs)分散在亲水性载体中,达到增溶的目的。近年来,由于技术的进步,无定型固体分散体(amorphous solid dispersion,ASD)受到学者广泛关注[5]。
相比其他制备固体分散体的方法(如喷雾干燥法、熔融法等),热熔挤出(hot-melt extrusion,HME)技术作为目前制备固体分散体提高药物溶解度和生物利用度的重要手段之一,具备可连续操作,不使用有机溶剂等优点,因此被认为是一种绿色环保的增溶技术[6]。通过混合、加热以及剪切力的作用,药物会以无定型或晶型的状态均匀分散在载体中,甚至达到分子水平的混合。
为了提高ITZ的体外溶出度,本实验采用HME技术,选取PEG6000/乙烯基己内酰胺/醋酸乙烯酯共聚物(Soluplus)、乙烯吡咯烷酮/醋酸乙烯酯(6∶4)共聚物(Kollidon VA64)、醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)和聚丙烯酸树脂(Eudragit EPO)为载体制备无定型固体分散体。同时用X射线粉末衍射(XRPD)、偏振光显微镜(PLM)和温度调制式差式扫描量热法(MT-DSC)分析所制备的无定形固体分散体的物理状态,考察影响溶出度的因素。
1 材 料
1.1 药品与试剂
伊曲康唑(纯度99.3%,上海珂华生物科技有限公司);斯皮仁诺胶囊(西安杨森制药有限公司);Soluplus(Sol)、Kollidon VA64(德国巴斯夫公司);HPMCAS(美国亚什兰集团公司);Eudragit EPO(EPO,德国赢创工业集团);乙腈、磷酸为色谱纯;其他试剂为市售分析纯。
1.2 仪 器
Pharmamini热熔挤出机;Nicolet 6700-NXR傅里叶红外光谱仪(美国赛默飞世尔有限公司);高效液相色谱仪(日本岛津公司,含LC-20ADXR高压泵,SPD-M20A二极管阵列检测器);Q2000差示扫描量热分析仪、Q5000热重分析仪(美国沃特世公司);XPR10电子天平(美国梅特勒-托利多公司);708-DS溶出仪(美国安捷伦科技有限公司);LV100PL偏振光显微镜(日本株式会社尼康);D8 Advance X射线粉末衍射仪(美国布鲁克道尔顿公司)。
2 方法与结果
2.1 药物与辅料处方前研究
2.1.1 药物与辅料热性质考察 分别称取ITZ、Soluplus、Kollidon VA64、HPMCAS和 Eudragit EPO约5 mg,置于热重分析仪(TGA)和差示扫描量热仪(DSC)盘中,以空盘为参比物,升温速度10℃/min,升温范围30~300℃,氮气环境下测定其热曲线。结果见图1和2。TGA图显示,药物在30~300℃范围内稳定,温度高于330℃时开始分解。4种辅料在30~200℃范围均稳定,HPMCAS和Eudragit EPO在约210℃开始降解,Soluplus和Kollidon VA64在250℃附近开始降解[7]。DSC图显示药物在第1次加热到167℃附近有一明显的吸热峰,此温度对应药物的熔点。随后冷却到室温后再加热,在59℃出现玻璃态转化温度(Tg),75℃和90℃出现的峰与药物的分子结构和玻璃态的手性向列中间相有关[8],具体机制仍不十分明确。
2.1.2 药物溶解度测定 色谱条件与系统适用性实验:参照现有方法[9],使用 Ascentis Express C18色谱柱(4.6 mm×100 mm,2.7μm),流动相 A:磷酸调至pH 2.5的0.1%三乙胺溶液-四氢呋喃(95∶5);流动相 B:乙腈,等比例进行等度洗脱,流速1.0 mL/min,柱温 30℃,进样体积 5μL,检测波长259 nm。
精密称取ITZ原料药约10 mg,置100 mL量瓶中,加稀释剂(甲醇-四氢呋喃,1∶1)20 mL,超声使其溶解,继续稀释至刻度,摇匀。精密量取适量,用稀释剂稀释,制成质量浓度分别为0.985,4.925,9.985,49.25,99.85μg/mL的系列标准溶液,同法测定。以峰面积对质量浓度进行线性回归,得回归方程为A=10 228c+5 091.8(r2=1,n=5),线性范围0.985~99.85μg/mL。
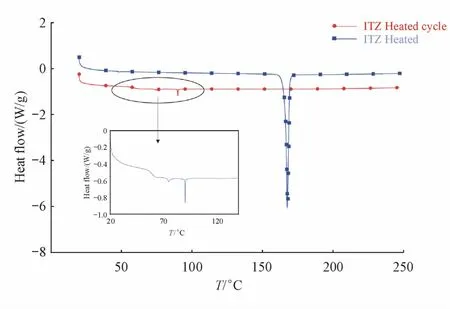
Figure 1 Differential scanning calorimetry(DSC)curves of itraconazole(ITZ)

Figure 2 Thermo-gravimetric analyzer(TGA)curves of ITZ and excipients
考察 ITZ在 SGF(0.1 mol/L HCL)、磷酸盐缓冲液(pH 6.8)和水中的溶解度。分别配置上述溶液,并取适量加入过量ITZ,密封后放入震荡混悬仪,温度控制在(37±1)℃,转速700 r/min,充分振摇使药物溶解并达到饱和。24 h后取2 mL于离心管中,14 000 r/min离心5 min,取上清液重复上步操作进行二次离心,再取上清液经稀释剂稀释后用HPLC分析,将峰面积带入标准曲线方程,计算溶解度。结果显示,ITZ在 SGF(0.1 mol/L HCL)中溶解度为(5.9±2.2)μg/mL,而在水和磷酸盐缓冲液(pH 6.8)中均低于1μg/mL,结果与文献报道相似[2]。
2.2 载体筛选
2.2.1 溶解度参数及玻璃态转化温度 溶解度参数可以通过确定物质的内聚能密度(CED)来预测他们之间的相容性。一般来说,药物与载体的溶解度参数之差Δδ<7.0 MPa1/2时,二者相容性良好。当 Δδ>10.0 MPa1/2时,二者不能相容[10]。Hansen将液态的内聚能视为色散力、极性力和氢键3种分子间作用力的贡献之和,其表达见公式(1)

式(1)中 δd、δp、δI分别是色散溶解度参数、极性溶解度参数和氢键溶解度参数。现经查阅文献和采用化学基团贡献法计算出ITZ和载体的溶解度参数(表1)。玻璃态转化温度(Tg)是HME制备固体分散体时载体选择的重要参数之一。载体的Tg一般应介于50~130℃之间,保证常温下载体为固体状态,同时防止HME过程的温度过高。除此之外,药物与载体的降解温度要比Tg高50℃以上,防止加热过程中发生降解[7]。

Table 1 Solubility parameters,T g and T deg of ITZ and different carries
2.2.2 MT-DSC法筛选载体 MT-DSC法可以初步模拟HME加热熔融的过程,通过加热-冷却-加热的过程判断经过一次加热后药物与载体是否形成均相,因此可以作为快速筛选载体的方法[11]。按照不同载药量设置3个水平,质量分数分别为10%、30%和50%(以下均为质量分数)。称取二元混合样品约5 mg至铝盘中,在30℃下恒温3 min,再以2℃/min的速率加热到190℃,调制温度振幅为1℃/min,见图3和4。载药量为10%的情况下,没有出现明显的吸热峰,表明所有载体与ITZ均形成ASD。载药量提高至30%时,ITZ与Eudragit EPO组出现明显吸热峰,其他组仍未出现。再次提高载药量至50%,所有组均出现明显吸热峰。上述结果说明对于所选4种载体,10%均为安全的载药量,同时ITZ与Eudragit EPO的相容性较其他3种载体稍差。因此,本实验主要探究载药量为30%和50%时ITZ与载体的关系。
2.3 HME技术制备固体分散体
一般情况下,挤出温度越高,药物熔融更加完全,在载体中的分散程度越大;与此同时,温度越高载体的黏度越低,流动性越大,越有利于药物分散在载体中,因此升高热熔挤出的操作温度,有利于增加药物的溶出[12]。但是,另一方面,随着挤出温度升高,药物受热降解的可能性也在增加。根据图1和图2结果所示,ITZ与4种载体的降解温度均超过200℃,ITZ熔点为167℃,因此设定挤出温度为170℃,确保不发生降解的前提下使药物达到熔融状态,与载体充分混合。HME螺杆的转速通过影响药物的保留时间对药物的物理状态产生影响。当转速较低时,药物和载体在特定挤出温度下暴露时间过长,可能会发生降解。与此相反,如果转速过高,螺杆产生的机械能和热能也可能导致降解[13]。因此,本实验选取50 r/min作为适中的挤出转速。
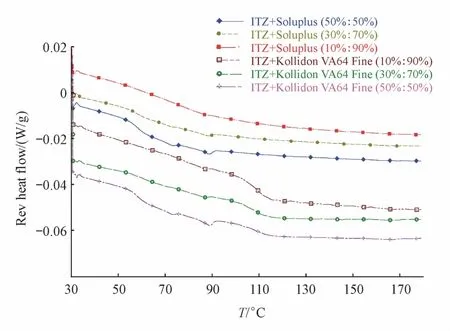
Figure 3 Reversible curves of binary mixtures of ITZ and carriers(Soluplus and Kollidon VA64)by modulated temperature-differential scanning calorimetry(MT-DSC)

Figure 4 Reversible curves of binary mixtures of ITZ and carriers(HPMCASand Eudragit EPO)by MT-DSC
与MT-DSC法相比,通过HME法制备ASD的过程中药物与载体在双螺杆剪切力的作用下更容易形成均相,因此结合图3和图4的结果,将载药量设置为30%和50%。按照上述比例,精确称取适量ITZ粉末与载体,加入玛瑙研钵中进行研磨混合。预先将HME恒温至170℃,将混合物通过HME加料口匀速加入,调整螺杆转速为50 r/min,观察扭矩数值,待其稳定后收集挤出物,冷却至室温,粉碎,过60目筛网待测。
2.4 物理表征
2.4.1 温度调制式差示扫描量热法 MT-DSC谱图的吸热和放热峰提供了相转变、物理状态和混合性等信息。玻璃态转化温度的出现或药物熔点的消失通常说明无定型体系的形成,反之,放热峰的出现一般说明发生了重结晶。对于二元混合物,单一的玻璃态转化温度取代原本两种物质的熔点说明他们从分子水平上形成了无定型固体分散体[10]。本实验采用MT-DSC法分析药物在固体分散体中的状态,分别称取约5 mg粉末至铝盘中,方法同“2.2.2”项,结果见图5和图6。当ITZ载药量为30%时,药物原本的熔点峰消失,4种载体中都没有出现明显的吸热峰,Tg分别出现在56℃、95℃、93℃和45℃。载药量为50%时,Soluplus和Kollidon VA64制备的固体分散体在90℃和158℃出现较小的吸热峰,90℃为ITZ在玻璃态下的手性向列相吸热峰,158℃为药物在与载体混合后的熔点,说明ITZ与 Soluplus和 Kollidon VA64在1∶1的比例下进行热熔挤出不能完全达到ASD的状态,仍有部分ITZ以晶体和中间相存在。除此之外,ITZ与HPMCAS制备的SD在90℃出现吸热峰,可能是部分药物分子以中间相存在。ITZ与载体Eudragit EPO制备的SD会在115℃发生重结晶,在167℃出现药物熔点峰,这可能是由于ITZ与Eudragit EPO的相容性较差,导致载体的抑晶效果较弱。而在载体含量为70%时,重结晶和熔化的焓明显小于50%载药量的样品,说明Eudragit EPO的含量与抑制ITZ重结晶的能力呈正相关。
2.4.2 偏振光显微镜 取适量固体分散体放置在载玻片上,滴加适量硅油进行分散。将制好的样片放置在载物台上,使用10×10倍放大倍数观察并拍摄样品,结果见图7。图7A-D中没有出现明显的双折射现象,可能由于ITZ与Soluplus和Kolli-don VA64混合性较好,故固体分散体处于无定型状态。图7-E、F中部分出现部分双折射现象,可能和辅料HPMCAS结晶状态有关。图7-G、H中部分出现双折射现象,可认为固体分散体中仍有部分未达到无定型状态。在降低载药量后,图7-H出现明显变化,无定型区域增多,双折射减少。

Figure 5 MT-DSC of ITZ solid dispersion via heat mect extrusion(HME)in different proportion

Figure 6 MT-DSC of ITZ solid dispersion with Eudragit EPO via HME in different proportion

Figure 7 Polarized lightmicroscope(PLM)images of ITZ HMEswhen the ratio of ITZ and carrierswere 50∶50 and 30∶70,respectivelyA:ITZ+Soluplus(50%∶50%);B:ITZ+Soluplus(30%∶70%);C:ITZ+Kollidon VA64(50%∶50%);D:ITZ+Kollidon VA64(30%∶70%);E:ITZ+HPMCAS(50%∶50%);F:ITZ+HPMCAS(30%∶70%);G:ITZ+EPO(50%∶50%);H:ITZ+EPO(30%∶70%)
2.4.3 X射线粉末衍射 使用XRPD对ITZ在SD中的分散状态进行扫描分析。工作条件:铜靶,管压40 kV,管流40 mA,扫描速度为10°min,扫描范围3°<2θ<40°。如图8所示,与ITZ原料相比,4种载体制备的SD均没有明显的衍射峰出现。这个结果与文献报道一致[14],说明大部分药物以非晶态存在。
2.4.4 傅里叶变换红外光谱 通过测定红外光谱判断固体分散体中ITZ与载体的相互作用,结果见图9。ITZ的羰基峰位在1 696 cm-1,为一尖锐峰;Soluplus的两个羰基峰位分别在1 732和1 624 cm-1;Eudragit EPO的羰基峰位在 1 723 cm-1。ITZ与Soluplus物理混合物的红外吸收图谱是分别图谱的简单叠加,而固体分散体的羰基峰减弱并且展宽向高波数端移动,可能是由于ITZ与Soluplus发生氢键相互作用。ITZ在2 821 cm-1处的烷基在固体分散体中减弱,可能是受到氢键外的其它作用力的影响。此外,受到Eudragit EPO中羰基的影响,ITZ-Eudragit EPO固体分散体的红外谱图未观察到明显变化。
2.5 初步体外评价
2.5.1 体外溶出度考察 精密称取ITZ原料药或挤出物粉末适量(含ITZ 50 mg),参照溶出度测定法[《中华人民共和国药典》(2015年版)四部通则0931第二法],以0.1mol/L盐酸溶液900 mL为溶出介质,转速为 75 r/min,温度为(37±0.5)℃,依法操作,于 5,10,30,60,90,120,180 min取液2 mL,0.45μm微孔滤膜滤过,取续滤液,适当稀释后作为供试品溶液。使用HPLC测定259 nm波长处供试品的峰面积,按标准曲线(参见“2.1.2”项),计算各时间点的累积溶出百分度。
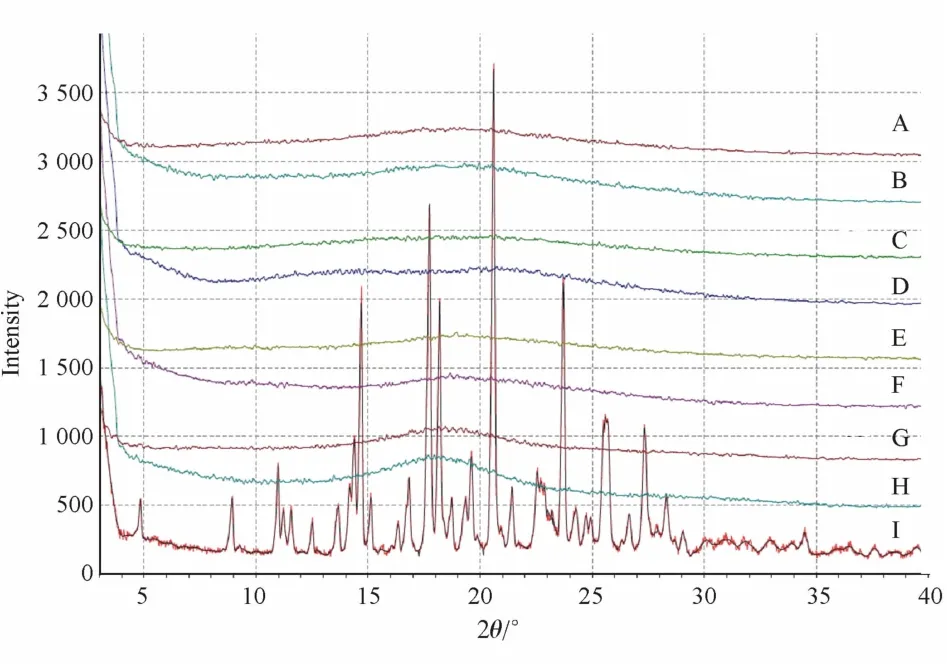
Figure 8 XRPD image of ITZ and extrudates by HMEA:ITZ+Sol(50%∶50%);B:ITZ+Sol(30%∶70%);C:ITZ+VA64(50%∶50%);D:ITZ+VA64(30%∶70%);E:ITZ+HPMCAS(50%∶50%);F:ITZ+HPMCAS(30%∶70%);G:ITZ+EPO(50%∶50%);H:ITZ+EPO(30%∶70%);I ITZ Raw
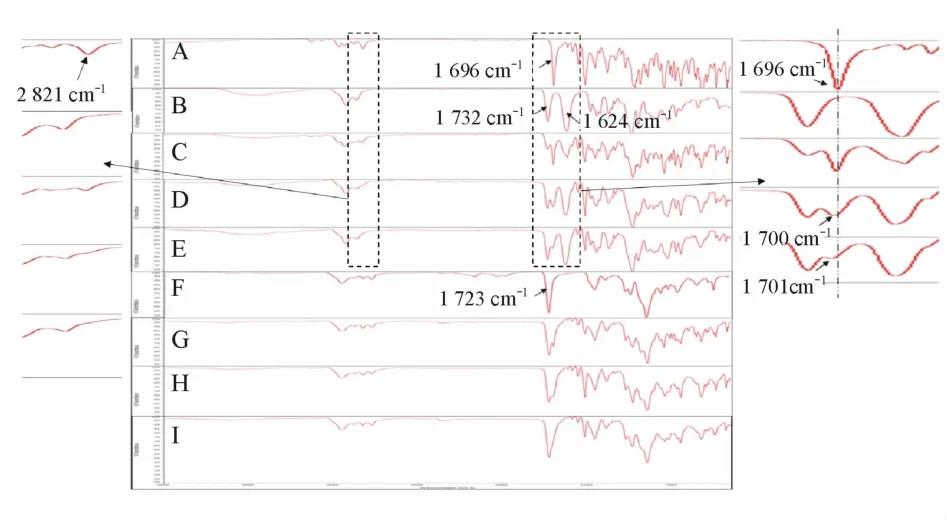
Figure 9 FT-IR spectroscopy of ITZ and extrudates by HMEA:ITZ raw;BSoluplus;C ITZ+Sol PMs(PhysicalMixtures);D:ITZ+Sol(50%∶50%);E:ITZ+Sol(30%∶70%);F:Eudragit EPO;G:ITZ+Eud PMs;H:ITZ+Eud(50%∶50%);I:ITZ+Eud(30%∶70%)
考察HME制备的SD的溶出度,并与原研制剂斯皮仁诺进行比较,结果如图10、11所示。对于两种不同的载药量,ITZ与 Soluplus和 Eudragit EPO制备的SD在SGF(0.1moL/LHCL)中溶出迅速,30 min累计溶出量达到90%,并且一直保持该状态未见析出。与此相比,市售原研制剂斯皮仁诺在90 min累计溶出量达到90%。Kollidon VA64组累计溶出量均未超过70%。HPMCAS组基本不溶,可能是HPMCAS为肠溶材料所致。
2.5.2 动力学溶解度曲线 ASD由于具有较高的能量,在溶液中有自发结晶的趋势[15]。因此通过测定SD在过饱和溶液中的动力学溶解度曲线,达到进一步筛选载体的目的。分别称取过量的ITZ固体分散体于瓶中,加入0.1 mol/L盐酸适量,用磁力搅拌器以200 r/min的速率在室温下搅拌24 h。分别在5,15,60,240,480,720,1 440 min取样过滤稀释,使用HPLC进行测定,并绘制溶解度与时间的变化曲线。如图11所示,ITZ与Soluplus制备的ASD达到了更高的过饱和度,并且在24 h内未发生明显重结晶,平衡溶解度明显高于其他载体。Kollidon VA64和Eudragit EPO组在平衡一段时间后均析出,而HPMCAS组以一定速率持续缓慢溶解。

Figure 10 Dissolution profiles of ITZ SD(50∶50)via HME with different carriers(¯x±s,n=3)

Figure 11 Dissolution profiles of ITZ SD(30∶70)via HME with different carriers,n=3)
2.6 初步稳定性评价
分别取制备的ITZ固体分散体适量于玻璃皿中,敞口条件放置在40℃,75%RH条件下,于0、30 d取样,通过XRPD判断其是否发生转晶。结果显示,此条件下放置30 d后,各组均未出现明显的晶体衍射峰。其中 B、C、D组在4°(2θ)出现的峰推测是中间相的特征峰[7],A组未出现可能是由于ITZ与Soluplus相容性较好,仍处于无定型状态。含量及有关物质检测方法:取固体分散体细粉适量(约相当于ITZ 25mg),置25mL量瓶中,加稀释剂超声溶解,稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液(1 mg/mL),精密量取供试品适量,用稀释剂稀释制成0.1 mg/mL的溶液,作为对照溶液,参照2.1.2中色谱条件,取对照溶液5μL注入液相色谱仪,调节检查灵敏度,使主成分色谱峰的峰高为满程量的10%~25%;再取供试品溶液与对照溶液各5μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3倍。含量测定:按照高效液相色谱法[《中华人民共和国药典》(2015年版)四部通则0512]测定。结果显示,在40℃,75%RH条件下放置30 d后,固体分散体的含量与溶出度无明显变化,未观察到明显杂质,ITZ-Soluplus和ITZ-EPO组均在30 min内溶出度达到90%以上;含量为原含量的98%~102%。

Figure 12 Kinetic solubility curve of ITZ extrudates by HME

Figure13 XRPD image ofextrudates in 40℃/75%RH condition for onemonthA:ITZ+Sol(30%∶70%);B:ITZ+Sol(50%∶50%);C:ITZ+EPO(50%∶50%);D:ITZ+EPO(30%∶70%);E:ITZ Raw
3 讨 论
HME技术是提高难溶性药物溶出度的有效方法之一。通过将药物与载体加热至熔融状态,在剪切力的作用下制备无定型固体分散体。其中载体对药物起到增溶且抑制重结晶的作用,因此载体的选择十分关键。根据体外溶出度的结果,Soluplus与Eudragit EPO均可以显著提高ITZ的溶出度,并且溶出速率较市售原研药斯皮仁诺有所提高。
分别对样品进行物理表征发现,ITZ与Soluplus在170℃下得到的固体分散体以无定型存在,而在Eudragit EPO中为介于无定型与晶型之间的向列中间相。可以认为ITZ与Eudragit EPO只发生了部分的分子层面的互溶,这个结果与溶解度参数预计的结果相符合。
动力学溶解度的结果证明,无定型固体分散体中Soluplus的增溶效果优于Eudragit EPO,同时也具有抑制ITZ在溶液中重结晶的作用。而ITZ与Eudragit EPO制备的固体分散体在溶液中析出,可能是由于Eudragit EPO的抑晶效果弱于Soluplus,也可能是由于分散的ITZ以中间相存在,更容易向晶型发生转变。综上所述,ITZ以无定型态与Soluplus达到分子层面互溶是其溶出度增加的主要因素。
目前通过HME制备的上市药物大部分需要制成其他剂型,因此将实验中制备的ASD放置在40℃,75%RH的条件下30 d,未发现有ITZ发生重结晶现象,说明其物理稳定性较好,长期稳定性仍然有待考察。此外,关于ITZ的无定型与中间相的研究仍有待深入。
[1] Bin C,Nong LL.Advances in itraconazole[J].Medical Recapitulate(医学综述),2010,16(8):1234-1236.
[2] Zhong Y,Jing G,Tian B,et al.Supersaturation induced by Itraconazole/Soluplus®micelles provided high GIabsorptionin vivo[J].Asian JPharm Sci,2016,11(2):255-264.
[3] Choi J S.Enhanced stability and solubility of pH-dependent drug,telmisartan achieved by solid dispersion[J].J Drug Deliv Sci Technol,2017,37:194-203.
[4] Chen YH,Wang QS,Ping QN,et al.Preparation of iguratimod solid dispersion via hot-melt extrusion and investigation of factors affecting dissolution profile[J].Chin Pharm J(中国药学杂志),2013,17:1272-1278.
[5] Chaudhari SP,Dugar RP.Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs[J].JDrug Deliv Sci Technol,2017,41:68-77.
[6] Ashour EA,Majumdar S,Alsheteli A,et al.Hotmelt extrusion as an approach to improve solubility,permeability and oral absorption of a psychoactive natural product,piperine[J].J Pharm Pharmacol,2016,68(8):989-998.
[7] Thiry J,Lebrun P,Vinassa C,etal.Continuous production of itraconazole-based solid dispersions by hotmelt extrusion:preformulation,optimization and design space determination[J].Int J Pharm,2016,515(1):114-124.
[8] Six K,Verreck G,Peeters J,etal.Investigation of thermal properties of glassy itraconazole:identification of a monotropic mesophase[J].Thermochim Acta,2001,376(2):175-181.
[9] Roy C,Chakrabarty J,Patel HB.Development and validation of a stability indicating binary RP-UPLCmethod for determination of itraconazole in capsules dosage form[J].Int JAnalyt Bioanalytical Chem,2012,2(3):165-174.
[10]Sarode AL,Sandhu H,Shah N,et al.Hotmelt extrusion(HME)for amorphous solid dispersions:predictive tools for processing and impact of drug-polymer interactions on supersaturation[J].Eur JPharm Sci,2013,48(3):371-384.
[11]Fule R,Meer T,Sav A,et al.Solubility and dissolution rate enhancement of lumefantrine using hotmelt extrusion technology with physicochemical characterisation[J].J Pharm Investig,2013,43(4):305-321.
[12]LaFountaine JS,McGinity JW,Williams RO.Challenges and strategies in thermal processing of amorphous solid dispersions:a review[J].AAPSPharmSciTech,2016,17(1):43-55.
[13]Thiry J,Krier F,Evrard B.A review of pharmaceutical extrusion:critical process parameters and scaling-up[J].Int J Pharm,2015,479(1):227-240.
[14]Hughey JR,Keen JM,Miller DA,et al.Preparation and characterization of fusion processed solid dispersions containing a viscous thermally labile polymeric carrier[J].Int J Pharm,2012,438(1):11-19.
[15]Van den Mooter G.The use of amorphous solid dispersions:A formulation strategy to overcome poor solubility and dissolution rate[J].Drug Discovery Today,2012,9(2):e79-e85.
