盐酸缬更昔洛韦的合成路线优化
2018-05-02汪小兵长江大学化学与环境工程学院湖北荆州434023荆楚理工学院湖北省荆门医药工业技术研究院湖北荆门448000
汪小兵 (长江大学化学与环境工程学院,湖北荆州434023 荆楚理工学院湖北省荆门医药工业技术研究院,湖北荆门448000)
尹先清 (长江大学化学与环境工程学院,湖北 荆州 434023)
许方亮,李俊,李立威 (荆楚理工学院湖北省荆门医药工业技术研究院,湖北 荆门 448000)
盐酸缬更昔洛韦,化学名为(S)-2-氨基-3-甲基丁酸 (R,S)-2-[(2-氨基-6-氧代-1,6-二氢-9H-嘌呤-9-基)甲氧基]-3-羟基丙酯盐酸盐,是瑞士罗氏公司研制开发的抗病毒药物更昔洛韦的前体药物,2001年3月获美国FDA许可,2001年5月首次在美国上市,是治疗获得性免疫缺陷综合征(AIDS)病人发生的巨细胞病毒(CMV)视网膜炎的第1个口服使用的药物,在2003年5月扩大了其适应症,用于预防和治疗器官移植者继发CMV感染[1~4]。
关于盐酸缬更昔洛韦的合成文献报道主要有3种方案,第1种方案是以更昔洛韦为起始原料[5,6],分别有3种方法:①更昔洛韦直接与氨基被保护的缬氨酸成双酯[7,8],再选择性脱去一个羟基上的缬氨酸,最后脱保护得盐酸缬更昔洛韦。路线中选择性水解得单酯条件苛刻,单双酯分离困难,收率较低。②先保护更昔洛韦的一个羟基[9,10],然后再经酯化、脱保护得盐酸缬更昔洛韦。③将更昔洛韦2-位氨基和一个羟基保护起来[11,12],再经酯化、脱保护得目标产物盐酸缬更昔洛韦。选择性地添加或者脱保护基比较困难,产物难以纯化,不适于工业生产。第2种或第3种方案分别是以鸟嘌呤或者取代鸟嘌呤为起始原料[13,14],要经过缩合、氢化、水解、缩合及氢化几步得目标产物,路线长,总收率低,且2次氢化都用到H2/Pd-C体系,操作麻烦,原料成本高,不利于工业化生产。
为此,笔者参考第1种方案中的方法②,设计了一条简便合成盐酸缬更昔洛韦(目标产物(1))的路线(见图1),该路线以单乙酰更昔洛韦(中间体(2))为起始原料,和叔丁氧羰基-L-缬氨酸在羰基二咪唑(CDI)和三乙胺(TEA)的作用下发生酯化反应生成单乙酰叔丁氧羰基-L-缬氨酸更昔洛韦,然后先在三氟乙酸(TFA)的二氯甲烷溶液中脱去氨基上的叔丁氧羰基得到单乙酰L-缬氨酸更昔洛韦,再经过碳酸钾碱性条件水解脱去单羟基上的乙酰保护基得到缬氨酸更昔洛韦,最后再与氯化氢成盐得到目标产物(1),路线总收率为35.2%,产物纯度为98.9%,其中非对映异构体比例为49∶49,更昔洛韦含量为0.71%。合成的目标产物符合USP35标准规定(纯度不低于97%,更昔洛韦含量≤1.5%,非对映异构体比例在45∶55到55∶45之间)。
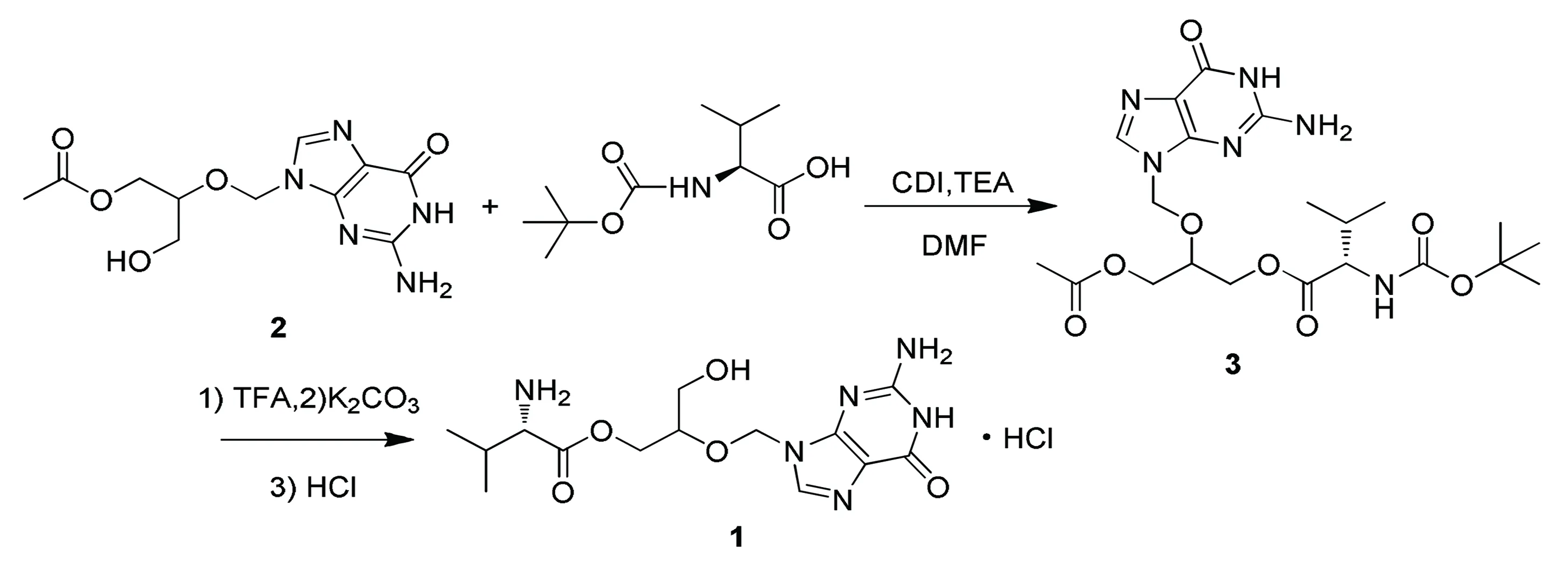
图1 目标产物(1)的合成路线
1 实验部分
1.1 主要仪器及试剂
仪器:AVANCEⅢHDD核磁共振仪-400MHz(TMS为内标,瑞士Bruker公司);Ultimate3000型高效液相色谱仪(美国赛默飞);DF101S型集热式恒温磁力搅拌器(巩义市予华)。RE52CS型旋转蒸发仪(上海亚荣);Accurate-Mass O-TOF 型LC /MS 质谱(Agilent)。
试剂:单乙酰更昔洛韦(98%);叔丁氧羰基-L-缬氨酸(Boc-L-缬氨酸)(98%);羰基二咪唑(CDI)(98%);三乙胺(TEA)(98%);N,N-二甲基甲酰胺(DMF)、甲醇、乙酸乙酯、盐酸、二氯甲烷、碳酸钾(K2CO3)(AR,国药试剂)。
1.2 单乙酰-Boc-L-缬氨酸更昔洛韦(中间体(3))的合成
100ml三口烧瓶中加入Boc-L-缬氨酸(5.5g,25.2mmol)、CDI(3.3g,20.2mmol)和DMF(20ml),N2保护下低温搅拌反应1.0h后升温至40℃,然后再加入中间体(2)(5.0g,16.8mmol)和三乙胺(5.1g,50.4mmol),置换气体后继续反应3.0h,TLC监控反应进程。反应毕,反应液加入到去离子水(100ml)中,加入乙酸乙酯(20ml)搅拌10min,分出有机相,水相再用乙酸乙酯(10ml×2)萃取,合并有机相,水洗2次,无水硫酸镁干燥,过滤,滤液减压浓缩得固体,用甲醇重结晶得6.3g白色固体,收率为75.4%,HPLC纯度为97.2%[HPLC归一化法:色谱柱Diamonsil C18柱(250mm×4.6mm,5μm);流动相A: 用磷酸调节pH值为2.8的10mmol/L的磷酸二氢钾水溶液,B:甲醇,梯度洗脱(0→5min:B 8%,5→15min:B 8%→20%,15→30min:B 20%→70%,30→40min:B 70%),检测波长:254nm,流速:1.0ml,柱温:35℃]。ESI-MS(m/z):497.1[M+H]+;1HNMR(400MHz ,CD3OD)δ:7.88(s,1H,NCHN),5.55~5.62(m,NCH2O),4.29~4.37(m,1H,CHNH),4.16~4.23(m,2H,CH2),4.04~4.15(m,2H,CH2),3.98~4.01(m,1H,OCH),1.98~2.09(m,1H,CHCH3),1.94(d,J=4.0Hz,3H,OCCH3)1.45(s,9H,CCH3),0.90~0.95(m,6H,CHCH3)。
1.3 盐酸缬更昔洛韦的合成
在100ml单口烧瓶中加入中间体(3)(3.0g,6.0mmol)和15%的TFA二氯甲烷溶液(10ml),室温下搅拌2.0h,TLC监控反应进程。反应毕,减压浓缩得残余物,然后加入甲醇(15ml)和水(6.0ml)溶解后,再加入K2CO3(2.78g,20.2mmol)固体,40℃下搅拌2.0h后,将反应液加入去离子水(60ml)中,用1mol/L的盐酸调节pH值至6左右,加入二氯甲烷(15ml)搅拌,分出有机层,水层用二氯甲烷萃取(2×10ml),合并有机相,有机相用饱和氯化钠水溶液洗2次,无水硫酸镁干燥,过滤,向滤液中通入氯化氢气体使溶液pH值为2~3,再将二氯甲烷旋蒸浓缩至干,将残余物用异丙醇和水的混合溶剂重结晶,得目标产物1.1g,收率为46.3%,HPLC纯度为98.9%,其中更昔洛韦含量为0.71%,非对映异构体的比例为49∶49,符合USP35标准规定(更昔洛韦含量≤1.5%,非对映异构体比例在45∶55到55∶45之间,总纯度不低于97%)。ESI-MS(m/z):355.7[M+H]+;1HNMR(400MHz,D2O)δ:7.86(d,J=3.6Hz,1H,NCHN),5.55(dd,J=4.8Hz、11.6Hz,1H,NCH2O),5.45(dd,J=7.2Hz、12.0Hz,1H,NCH2O),4.21~4.26(m,1H,CH2OH),4.01~4.08(m,1H,CH2OH),3.94~4.01(m,1H,OCH),3.69(dt,J=4.0Hz、12.4Hz,1H,CHNH2),3.62(dd,J=4.8Hz、17.2Hz,1H,CH2OOC),3.57(dd,J=4.0Hz、8.0Hz,1H,CH2OOC),1.76~1.92(m,1H,CHCH3),0.70~0.80(m,6H,CH3)。
2 结果与讨论
在中间体(3)的合成中,文献报道[7,10~12]采用的催化体系为二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP),该体系总是生成一种不易去除干净的二环己基脲(DCU)混在产物中,为产品纯化带来麻烦。而笔者采用的CDI和TEA体系可以避免这种麻烦,并且在合成过程中为了提高收率,对其反应条件进行了一定的优化。结果表明,当Boc-L-缬氨酸与单乙酰更昔洛韦的摩尔比为1.5,CDI与单乙酰更昔洛韦的摩尔比为1.2,反应温度为40℃时,该反应有较好的收率。
在第2步反应中,该研究将脱Boc保护基和脱单乙酰保护基通过先后加入TFA和K2CO3来实现。2步反应在“一锅煮”中完成,反应不再需要在加压釜中用贵重的Pd/C催化剂催化进行,降低了反应成本,并且使反应在本质上变得更加安全。
3 结语
在单乙酰-Boc-L-缬氨酸更昔洛韦的合成过程中,当反应条件为Boc-L-缬氨酸与中间体(2)的配比为1.5、CDI与中间体(2)的摩尔比为1.2、反应温度为40℃、在40℃下反应时长为3.0h时有较好的收率,收率可达75.4%,产物经HPLC检测的纯度为97.2%。在最后一步的合成关键步骤中,通过“一锅煮”的方式先后脱去了氨基保护基和羟基保护基,缩短了反应步骤。合成路线总收率为35.2%,目标产物(1)纯度为98.9%,其中非对映异构体比例为49∶49,更昔洛韦含量为0.71%。合成的目标产物符合USP35标准规定。研究路线具有原料易得、步骤少、反应条件温和、操作简单等优点,适宜于工业生产。
[参考文献]
[1]Emery V C,Hassan-Walker A F.Focus on new drugs in development against human cytomegalovirus[J].Drugs,2002,62(13):1853~1858.
[2]Weiskittel P.Valganciclovir hydrochloride:a new antiviral agent[J].Nephrology Nursing Journal Journal of the American Nephrology Nurses Association,2003,30(1):93.
[3]Sugawara M,Huang W,Fei Y J, et al.Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2[J].Journal of Pharmaceutical Sciences,2000,89(6):781~789.
[4]钟倩.嘌呤类核苷类抗病毒药缬更昔洛韦[J].世界临床药物,2003,24(2):119~120.
[6]Reddy G M, Prasada Raju V V N K V, Satyanarayana K, et al.Alter-native synthesis of valganciclovir hydrochloride[J].Synthetic Communications,2013,43(3): 425~430.
[7]胥涛,屈博毅,海俐,等.盐酸缬更昔洛韦及其有关物质的合成[J].中国医药工业杂志,2014,45(5):409~448.
[8]Arzeno H B.Process for preparing a 2-(2-amino-1,6-dihydro-6-oxo-purine-9-yl) methoxy -1,3-propanediol valinate[P].US:5700936,1997-12-23.
[9]Arzeno H B,Humphreys E R, Roberts C R, et al.Process for preparing a 2-(2-amino-1,6-dihydro-6-oxopurin-9-yl)methoxy-1,3-propanediol derivative[P].US:5840890,1998-11-24.
[10]Handa,Kamat,Sivakumaran.An improved process for preparation of Valganciclovir starting from triacetyl ganciclovir[P].IN:2012CH01644,2014-03-07.
[11]赵士魁,马康,郭庆明,等.盐酸缬更昔洛韦的合成[J].中国医药工业杂志,2015,46 (2):120~122.
[12]Arzeno H B, Humphreys E R.Process for preparing a 2-(2- amino-1,6-dihydro -6-oxo-purin-9-yl)-methoxy-1,3-propanediol derivative[P].US:5756736, 1998-05-26.
[13]Dvorak Charles A,Wren Douglas L,Fisher Lawrence E, et al.Process for preparing a 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol derivative [P].US: 6040446,2000-03-21.
[14]Nestor J R,John J, Maag Hans.2-(2-Amino-1,6-dihydro-6-oxo-purin-9-y1) methoxy-l,3-pro- panediol derivative[P].US:5840891,1998-11-24.
