三个晶状体半脱位家系FBN1基因和ADAMTSL4基因突变研究
2018-05-02,
,
1.北京丰台医院眼科,北京 100071; 2.北京同仁医院眼科,北京 100730
先天性晶状体半脱位是由于晶状体悬韧带先天发育异常,使得悬韧带部分或全部缺损,引起对晶状体的悬挂力不平衡或丧失,导致晶状体离开正常的生理位置,引起视力下降,可合并高度近视或远视等。先天性晶状体半脱位是一种结缔组织疾病,具有明显的遗传倾向,可以常染色体显性或常染色体隐形遗传模式进行遗传。已经证实FBN1基因突变是先天性晶状体半脱位常染色体显性遗传的致病基因[1];而常染色体隐形遗传的先天性晶状体半脱位与ADAMTSL4基因突变相关[2]。本研究对三个先天性晶体半脱位家系进行致病基因的研究,发现这三个家系所有患者均存在FBN1基因不同位点的错义突变。现报道如下。
1 研究对象
选择2014年2月—2014年9月在北京同仁医院白内障中心就诊的三个先天性晶状体半脱位家系,包括14例患者,男6例,女8例,年龄4~62岁和3名正常家系成员。通过询问家系成员病史和家族史,眼科及其他相关科室协助会诊,依据2010年修订版Ghent的疾病分类学标准进行临床诊断[3]。眼科检查包括视力,裂隙灯显微镜检查有无晶状体、晶状体状态及脱位的范围等。X线检查患者脊柱及四肢骨骼,彩色超声心动图排除心脏主动脉瘤样扩张疾病。本研究遵循赫尔辛基宣言,所有患者均知情同意。
1.1 方法
分别采集Ⅰ号家系(图1)10例患者及1名表型正常个体外周静脉血5 mL;Ⅱ号家系(图2)3例患者及1名表型正常个体外周静脉血5 mL;Ⅲ号家系(图3)1例患者与1名表型正常个体外周静脉血5 mL。EDTA抗凝,使用康为世纪通用型柱式基因组提取试剂盒提取样本DNA。
1.2 高通量FBN1基因测序[4]
利用目标基因捕获技术(北京中科检验),采取设计好的基因捕获探针与基因组DNA文库混合,对FBN1基因外显子区域进行捕获。目标区域基因片段被杂交到探针上,进而通过生物素和链霉亲和素的磁珠结合被吸附到磁珠上,通过洗脱处理将非目标区域的DNA片段洗掉,得到目标基因。采用Covaris S2超声仪将样本进行超声片段化。末端补平:分别取75 μL片段化的DNA、End Repair Reaction Buffer 14 μL、End Repair Enzyme Mix 11 μL(Mygenostics公司),总体积100 μL。20 ℃孵育30 min。用Beckman Ampure beads 按1.8∶1 体积比例(beads:反应体积)对产物进行纯化。片段两端加A(A-Tailing):末端补平纯化洗脱32 μL DNA、A-Tailing Reaction Buffer 15 μL、A-Tailing Enzyme Mix 3 μL(Mygenostics公司),总体积50 μL反应体系,37 ℃孵育30 min。产物采用Beckman Ampure beads 按1.8∶1体积比例(beads:反应体积)进行纯化。连接adaptor:A-Tailing 产物纯化洗脱27 μL DNA、Ligation Reaction Buffer 40 μL 、Ligation Enzyme Mix 30 μL(Mygenostics公司),总体积 97 μL反应体系,25 ℃孵育10 min。产物采用Beckman Ampure beads 按1.8∶1体积比例(beads:反应体积)进行纯化。将1 μg已制备好的文库的与BL缓冲液和设计的探针混合(表述不清),95 ℃加热7 min,65 ℃加热2 min;加入23 mL预热至65 ℃的HY缓冲液,65 ℃杂交22 h。用500 μL 1×结合缓冲液冲洗50 μL磁珠3次,63 μL 2×结合缓冲液至杂交混合物中,转移至含有80 μL磁珠的试管中。旋转混匀1 h。用 WB1缓冲液室温清洗磁珠15 min。然后用洗脱缓冲液将结合的DNA洗脱下来。洗脱的DNA进行PCR反应,反应条件如下:98 ℃预变性30 min;98 ℃变性20 s,65 ℃退火30 s,72 ℃延伸30 s,共进行9个循环;最后72 ℃延伸5 min,4 ℃延伸10 s。PCR产物采用Beckman Ampure beads 按1.8∶1 体积比例(beads:反应体积)进行纯化。采用取3 μL制备好的文库样本,做1%的琼脂糖凝胶电泳实验,电泳结果片段在300~500 bp的区域条带。利用HiSeq 2000高通量测序。利用Trim-Galore程序过滤低质量的序列,保留读出质量大于20 μL和读取长度大于80 bp的序列,然后使用GATK软件包对质量值进行校准,利用CCDS、人基因组数据库(HG19)、dbSNP(v144)信息对SNP进行注释,注释完成的数据再进一步比对HGMD、ClinVar、OMIM、UniProt等数据库进行分析并且针对每个位点进行文献确认。
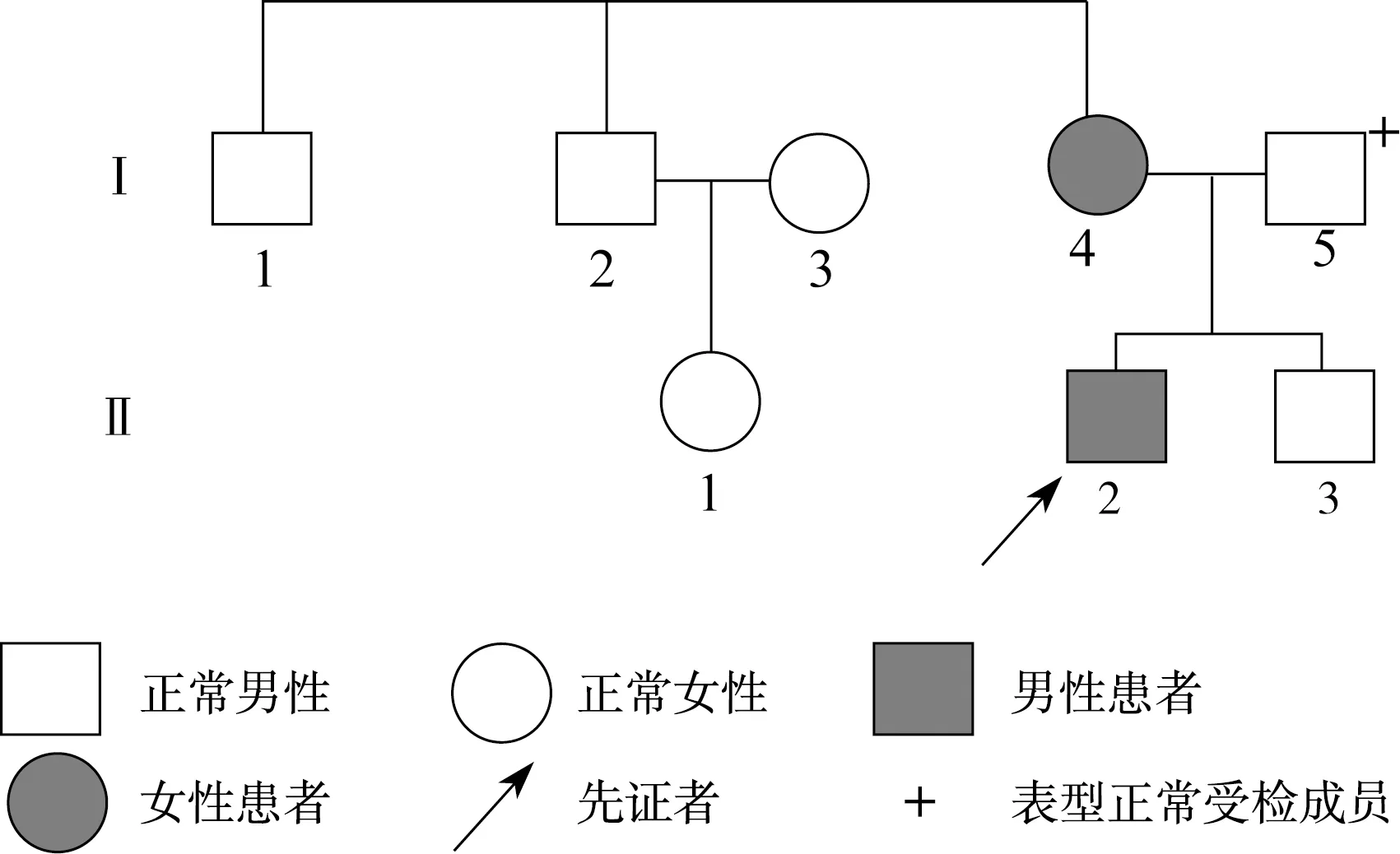
图2 Ⅱ号家系先天性晶体半脱位家系图Fig.2 The pedigree of congenital lens subluxation of the family Ⅱ
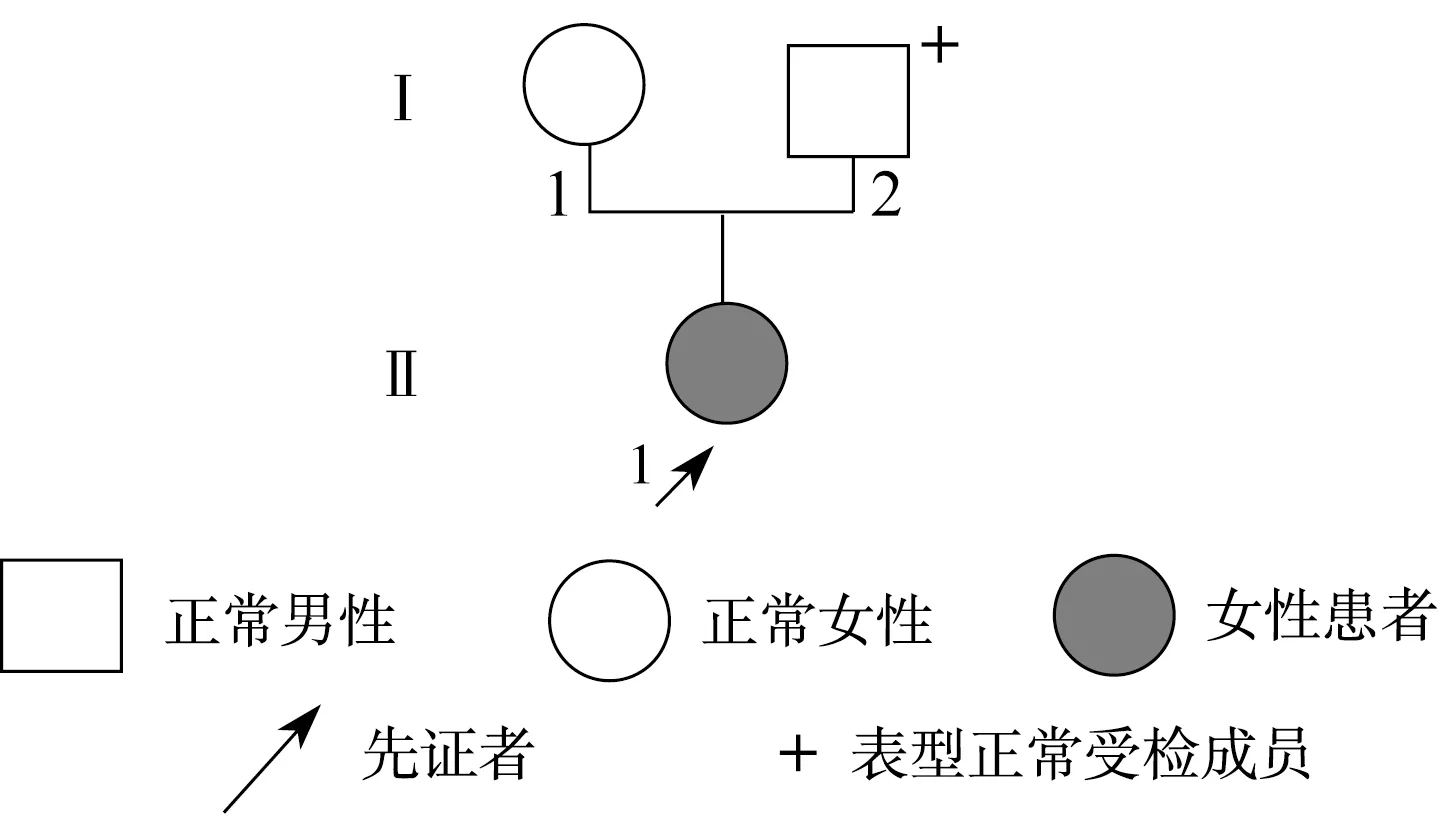
图3 Ⅲ号家系先天性晶体半脱位家系图Fig.3 The pedigree of congenital lens subluxation of the family Ⅲ
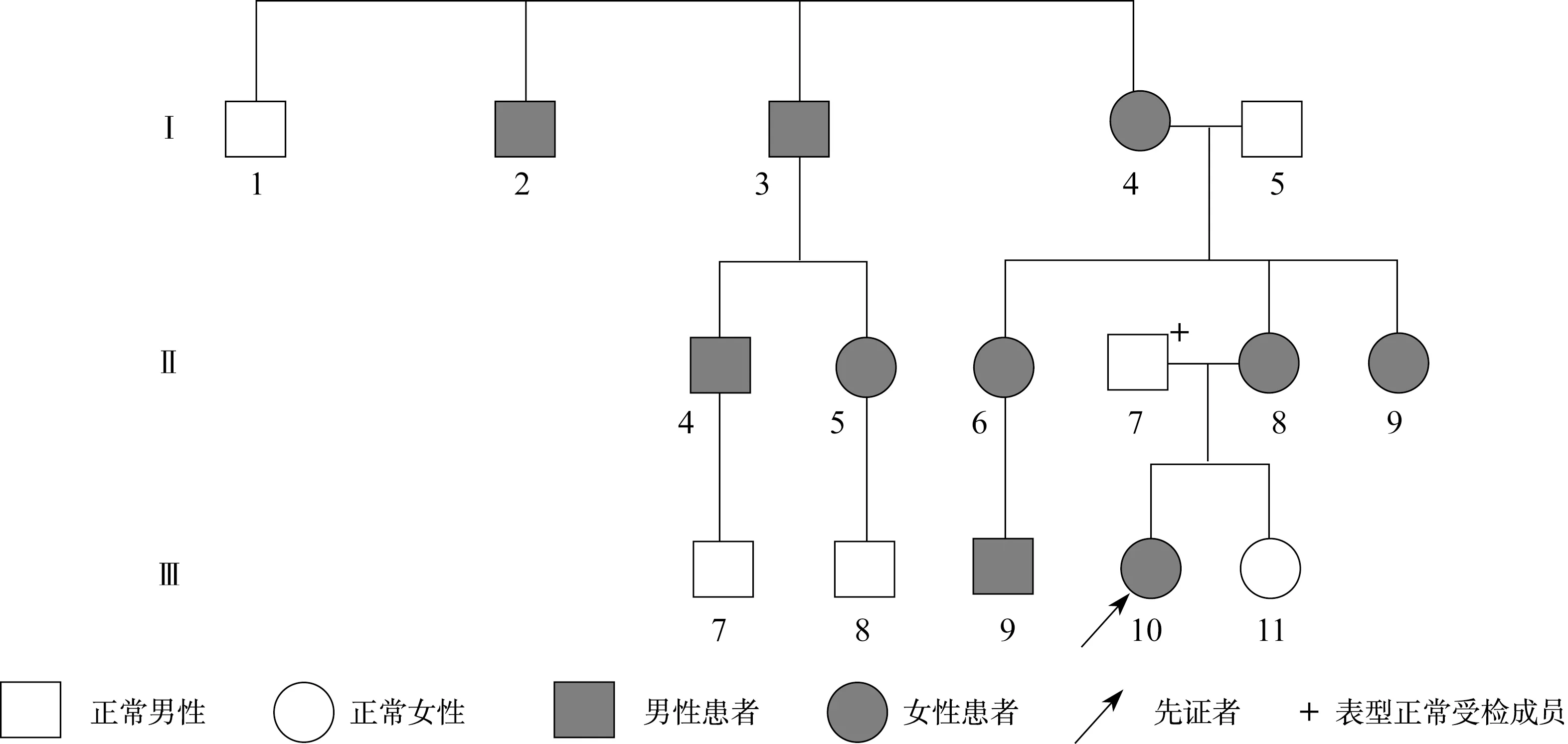
图1 Ⅰ号家系先天性晶体半脱位家系图Fig.1 The pedigree of congenital lens subluxation of the familyⅠ
1.3 Sanger法测序及验证
FBN1和ADAMTSL4基因编码区引物序列通过软件Oligo设计,由生工生物工程(上海)股份有限公司合成。FBN1和ADAMTSL4基因外显子扩增,取5 μL扩增产物与0.5 μL 10×loading buffer混匀后,点样到含Glod View的1%琼脂糖凝胶进行电泳验证,同时加入DNA Marker作为对照,在凝胶成像系统中鉴定扩增结果,通过对照DNA Marker 各片段位置和亮度来鉴定PCR产物片段的大小和浓度。采用试剂盒(AxyPrep DNA Gel Extraction Kit)柱法回收目标DNA进行凝胶电泳检测,测序产物进行扩增(使用试剂盒Big Dye Terminator V3.1 Cycle Sequencing Kit),测序产物进行纯化,应用ABI 3730XL自动测序仪完成测序。测序结果录入Chromas及DNAStar软件进行序列比对,经鉴定出的突变再进行反向测序进一步验证明确。
2 结果
2.1 FBN1基因
目标基因捕获结合高通量测序结果发现:Ⅰ号家系长期生活在我国河北,是一个常染色体显性遗传三代家系,成员共25名,患者10例,先证者为1例8岁患儿。家系所有患者的FBN1基因第38号外显子mRNA第4588位碱基均出现C→T杂合错义突变,即c.4588C>T,导致了第1530编码子编码的野生型精氨酸被突变型半胱氨酸替代,使第1530编码子发生了p.R1530C的突变(图4)。Ⅱ号家系长期生活在我国河北,是一个常染色体显性遗传三代家系,先证者为1例4岁患儿,3例患者的FBN1基因第40号外显子mRNA第4864碱基均出现T→C杂合错义突变,即c.4864T>C,导致了第1622编码子编码的野生型半胱氨酸被突变型精氨酸替代,使第1622编码子发生了p.C1622R的突变(图5)。Ⅲ号家系长期生活在我国山东,2例患者,其中1例已确诊患者病逝,先证者为1例28岁女性,其FBN1基因第16号外显子mRNA第1883位碱基出现G→A杂合错义突变,即c.1883G>A,导致了第628编码子编码的野生型半胱氨酸被突变型酪氨酸替代,使第628编码子发生了p.C628Y的突变(图6)。分别设计引物扩增后进行Sanger法测序,测序结果相同。家系正常个体,本地正常人数据库及1 000人基因数据库中均无此类突变。经Polyphen程序分析:Ⅰ号家系精氨酸被半胱氨酸替代后所得的PSIC值为0.998;Ⅱ号家系半胱氨酸被精氨酸替代后所得的PSIC值为0.999;Ⅲ号家系半胱氨酸被酪氨酸替代后所得的PSIC值为0.998,均提示野生型氨基酸被突变型氨基酸替代后,均引起FBN1蛋白结构及功能的破坏。
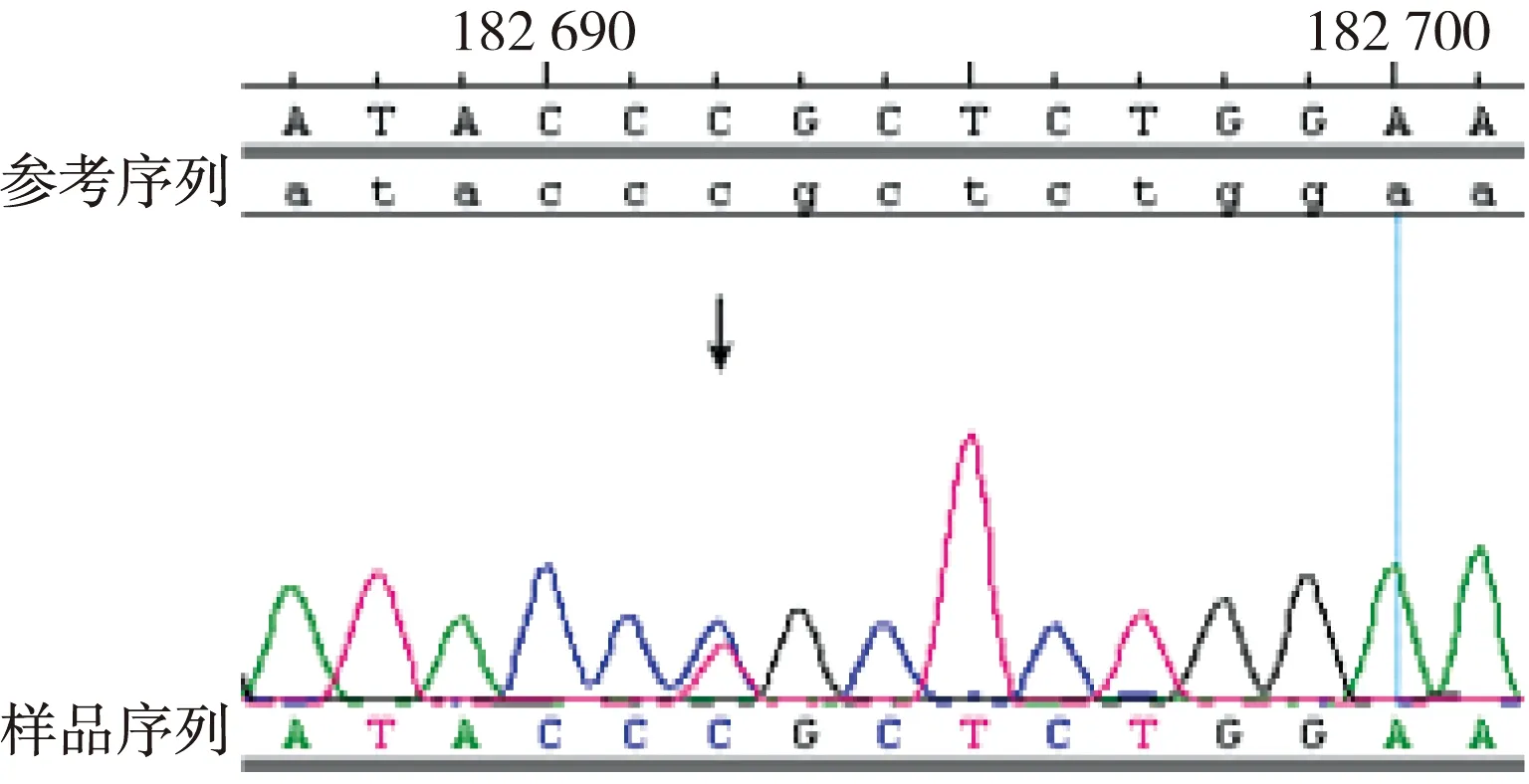
图4 突变型 Ⅰ号家系先证者的FBN1基因38号外显子部分测序结果(检测到突变:4588C>T)Fig.4 Identification of 4588C>T mutation in FNB1 exon 38
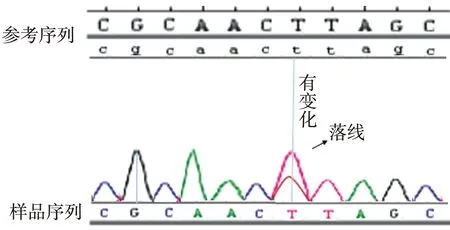
图5 突变型 Ⅱ号家系先证者的FBN1基因40号外显子部分测序结果(检测到突变:4864T>C)Fig.5 Identification of 4864T>C mutation in FNB1 exon 40
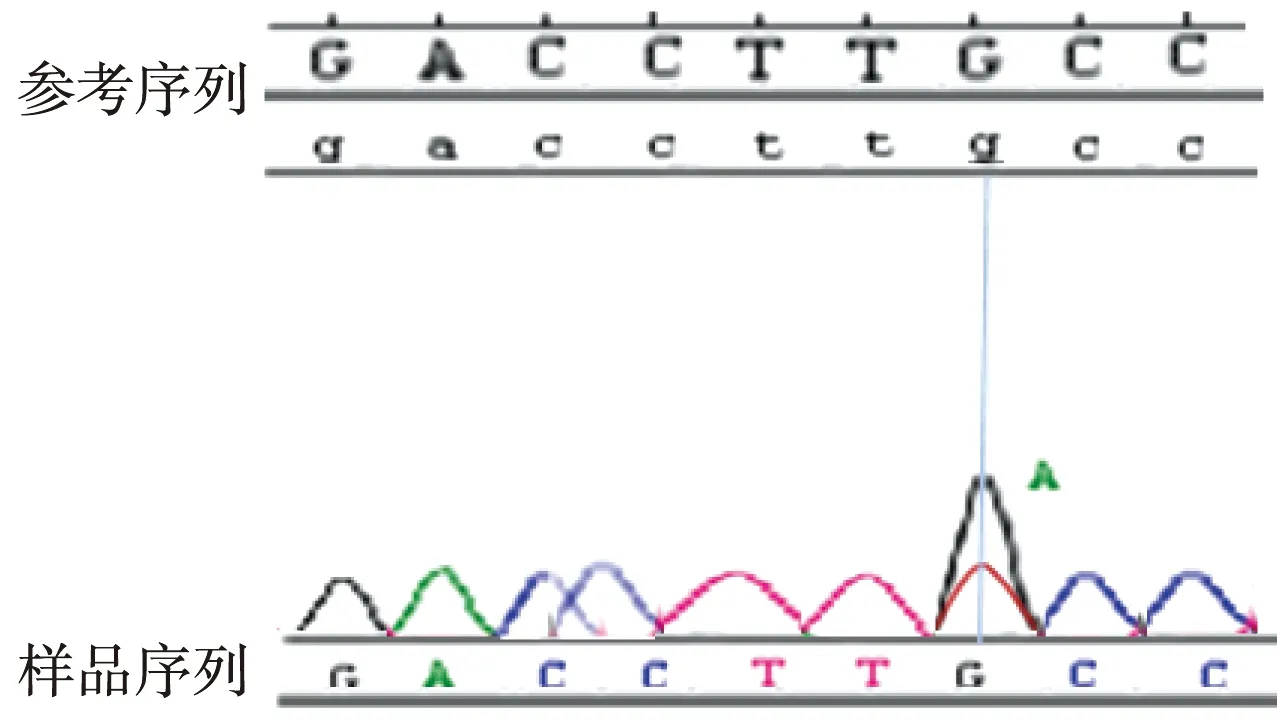
图6 突变型 Ⅲ号家系先证者的FBN1基因16号外显子部分测序结果(检测到突变:1883G>A)Fig.6 Identification of 1883G>A mutation in FNB1 exon 16
2.2 ADAMTSL4基因
Sanger法测序结果发现三个家系3例先证者ADAMTSL4基因均有不同外显子mRNA核酸位点的改变,但对蛋白的结构和功能没有改变,均为单核苷酸多态性改变(SNP),见表1。
3 讨 论
本研究中,使用了目标基因捕获技术联合高通量测序再结合Sager法验证基因检测结果的方法进行基因突变的筛查,这种方法能快速、准确、高通量、低成本地对单基因多个外显子或多个基因同时测序,大大提高了基因测序的效率、降低了成本,今后还可以用在大样本量的疾病已知基因检测中,使我们对分子病因学有更好的认识。
本研究中,确定了三个常染色体显性遗传的先天性晶状体半脱位家系。三个家系所有患者均表现为双眼晶状体向颞侧半脱位,经彩色多普勒超声心动图及骨骼X线检查,仅Ⅰ号家系3例患者身高超过1.90 cm,双手指细长,其他体征正常,彩色超声心动图未见异常。先天性晶状体半脱位与马凡综合征(MFS)均可以表现为常染色体显性遗传,在临床表型和基因型上均有一定程度的重叠性,有较高的外显性,同时又表现各异。先天性晶状体半脱位通常会有一些MFS的骨骼表现,但缺乏主动脉相关性疾病是其重要特征。依据修订版Ghent确诊马凡综合征(MFS)需要心脏、骨骼、眼三个系统受累,其中两个系统符合主要诊断标准,主动脉根部的扩张视为诊断的首要标准,更为强调结合心血管、骨骼系统、晶状体异位累及的程度、家族史和是否携带突变基因来综合诊断,而其他的临床表现如单纯性晶状体半脱位或合并骨骼系统的表现均不属于MFS。对候选基因FBN1及ADAMTSL4进行侧序,发现所有患者FBN1基因均存在杂合性错义突变,在蛋白层一级结构上分别导致了野生型的氨基酸残基被新的氨基酸残基所取代,而正常者未发现此突变。ADAMTSL4基因仅发现四个SNP位点,尚无相关致病性报道。FBN1基因定位于15号染色体15q21.1[5],全长约200 kb,是由65个外显子和47个内含子组成的复合多区域结构,其编码序列长约10 kb,由多个重复性的结构域构成。目前经UMD数据库检索已发现1 780多种基因突变,随机分布在整个FBN1基因中,没有明显的热点突变。FBN1蛋白在心血管系统、软骨、角膜、晶状体悬韧带以及皮肤、肾等组织广泛表达,是微纤维的重要组成部分[6],微纤维可调节组织中弹力蛋白之间及弹力蛋白与细胞外基质的相互作用,对组织可起到支撑作用。先天性晶状体脱位是由于晶状体悬韧带非对称性的过度松弛,在病理学检查时发现悬韧带纤维不仅在数量上减少,而且直径变细,形态不规则,与正常对照比较纤维无弹性,很容易断裂。在结构上,FBN1基因编码的FBN1蛋白前体,是由2 871个氨基酸组成,其包含6个结构域,其中类表皮样生长因子结构域(cb-EGF)与钙离子的结合可以保护FBN1蛋白的杆状结构,免于FBN1蛋白水解酶水解,钙结合域的突变及半胱氨酸残基的置换可引起FBN1蛋白前体结构域的变化,从而导致功能障碍[8],患者发生晶状体半脱位的概率相对较高。本研究三个家系患者:Ⅰ号家系患者FBN1基因38号外显子碱基杂合错义突变,导致第1530编码子编码的野生型精氨酸被突变型半胱氨酸替代,使其发生p.R1530C的突变;Ⅱ号家系患者40号外显子碱基杂合错义突变,导致了第1622编码子编码的野生型半胱氨酸被突变型精氨酸替代,使其发生p.C1622R的突变;Ⅲ号家系患者16号外显子碱基杂合错义突变,导致了第628编码子编码的野生型半胱氨酸被突变型酪氨酸替代,使其发生了p.C628Y的突变。本研究报道的这三个FBN1基因突变都位于原纤维蛋白-1钙结合结构域,半胱氨酸与精氨酸及酪氨酸的替代导致二硫键不能结合,cb-EGF的钙结合共有结构的空间构象发生改变,减弱了cb-EGF结合钙的能力,影响野生型蛋白高级结构的形成,干扰其功能。同时FBN1蛋白易于被降解,导致细胞外基质微纤维结构裂解,弹性纤维的稳定性下降,悬韧带的结构功能受到严重的影响。
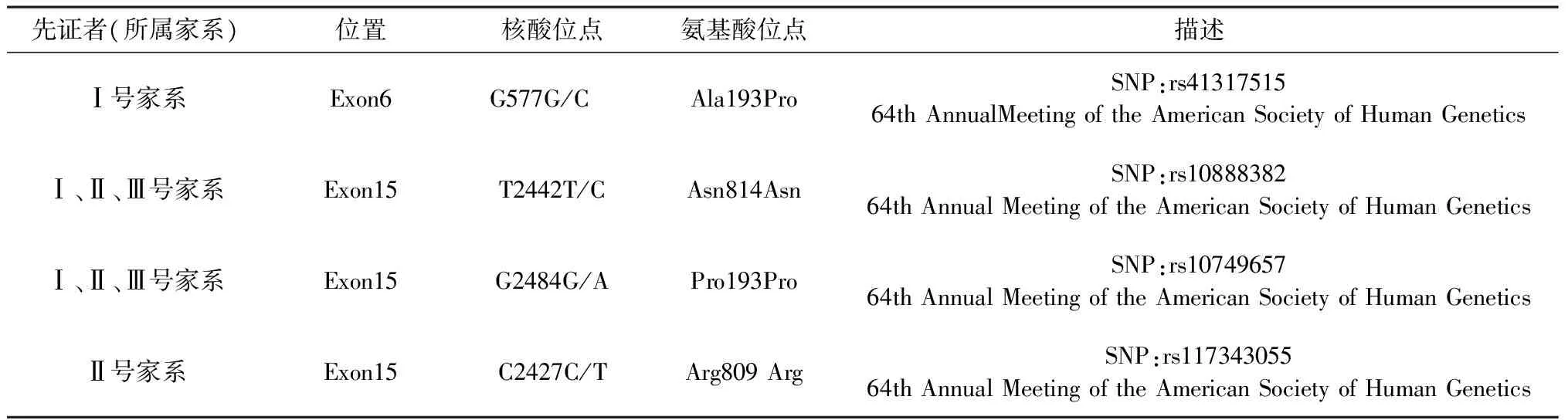
表1 3名先证者ADAMTSL4基因检测结果Tab.1 Detection of ADAMTSL4 gene in 3 probands
ADAMTSL4基因位于1号染色体(1q21.2),长约11.5 kb,包括全部19个外显子,mRNA为4 209 bp,由HEK293F细胞的转染表达。纯合及复合杂合ADAMTSL4基因突变是常染色体隐性遗传先天性晶状体半脱位的主要原因[9-12]。目前已报道了15种基因突变,大部分患者来自于欧洲民族,无义突变所导致的提前终止密码子及错义突变多见。ADAMTSL4蛋白质的功能及眼部定位尚未完全建立,有相关研究结果表示,mRNA和蛋白编码ADAMTSL4产物是一种分泌性糖蛋白,广泛分布于眼球,证明在虹膜、脉络膜、睫状体及视网膜分布。有学者曾报道在17例单纯晶状体半脱位患者FBN1和ADAMTSL4基因型和表型的不同特证中阐述[13]:ADAMTSL4基因突变的患者临床体征表现早,眼轴增长更明显。ADAMTSL4基因产物参与细胞外基质形成和内环境稳定,ADAMTSL4蛋白结合原纤维蛋白1的微纤维,可加速微纤维生物合成,在形成小带的过程中起到潜在的作用。本研究对家系中3例先证者的ADAMTSL4基因的检测结果经SSCP证实为单核苷酸多态性改变(SNP),而非基因突变。
通过蛋白的多重序列比对(采用DNASTAR软件中的negAlign程序)了解一个基因家族的基本特征,寻找序列的保守性。我们发现FNB1基因编码子1530、1622、628的野生型氨基酸在进化中高度保守,是斑马、鱼、鸡、狗、小鼠、大猩猩及人类原纤维蛋白-1蛋白(FBN1)功能域中的重要结构蛋白,此类蛋白的改变会引起物种基本特征的改变。
Polyphen程序(http://coo/enbl.elc/polyphen)是用来预测蛋白被替代后对其结构和功能产生损害作用的工具,一般认为PSIC 值越接近1.0,损害可能性越大,越接近0,损害可能性越小,通过polyphen程序分析进行预测突变意义。本文计算的三个家系的PSIC值结果分别是0.998、0.999、0.998,提示基因的突变损害了FBN1蛋白的功能,是基因的致病性突变,而非SNP。
本研究只对三个晶状体半脱位家系FBN1及ADAMTSL4两个候选基因进行了遗传学分析研究,样本量较少,因此FBN1及ADAMTSL4基因突变与先天性晶状体半脱位疾病的关系还有待进一步深入研究。
[1] 董松波,郑军,孙立忠.遗传综合征与主动脉瘤及夹层的关系[J].心肺血管病杂志,2012,31(5):523-526.
[2] Ahram D, Sato TS, Kohilan A, et al.A homozygous mutation inADAMTSL4 causes autosomal-recessive isolated ectopia lentis[J].Cell Press,2009,84(2):274-278.
[3] Loeys BL, Dietz HC, Braverman AC.The revised Ghent nosology for the Marfan syndrome[J].J Med Geneti,2010,47(7):476-485.
[4] Carvill GL, Heavin SB, Yendle SC.Targeted resequencing in epileptic encephalopathies identifies de novo mutations inCHD2 andSYNGAP1[J].Natur Genet,2013,45(7):825-830.
[5] Jaradat SA, Abujamous LA, Al-Hawamdeh AA, et al.Two novel mutations ofFBN1 in Jordanian patients with Marfan syndrome[J].Int J Clin Exp Med,2015,8(10):18786-18792.
[6] Collod-Beroud G, Le Bourdelles S, Ades L.Update of theUMD-FBN1 mutation database and creation of anFBN1 polymorphism database[J].Hum Mutat,2003,22(3):199-208.
[7] Zilberberg L, Phoon CK, Robertson I, et al.Genetic analysis of the contribution ofLTBP-3 to thoracic aneurysm in Marfan syndrome[J].Proc Natl Acad Sci U S A,2015,112(45):14012-14017.
[8] Yadin DA, Robertson IB, McNaught-Davis J, et al.Structure of the fibrillin-1 N-terminal domains suggests that heparan sulfate regulates the early stages of microfibril assembly[J].Structure,2013,21(10):1743-1756.
[9] Neuhann TM, Artelt J, Neuhann TF, et al.A homozygous microdeletion withinADAMTSL4 in patients with isolated ectopia lentis: evidence of a founder mutation[J].Invest Ophthalmol Vis Sci,2011,52(2):695-700.
[10] Aragon-Martin JA, Ahnood D, Charteris DG, et al.Role ofADAMTSL4 mutations inFBN1 mutation-negative ectopia lentis patients[J].Hum Mutat,2010,31(8):E1622-E1631.
[11] Greene VB, Stoetzel C, Pelletier V, et al.Confirmation ofADAMTSL4 mutations for autosomal recessive isolated bilateral ectopia lentis[J].Ophthalmic Genet,2010,31(1):47-51.
[12] Ahram D, Sato TS, Kohilan A, et al.A homozygous mutation inADAMTSL4 causes autosomal-recessive isolated ectopia lentis[J].Am J Hum Genet,2009,84(2):274-278.
[13] Chandra A, Aragon-Martin JA, Hughes K, et al.A genotype-phenotype comparison ofADAMTSL4 andFBN1 in isolated ectopia lentis[J].Invest Ophthalmol Vis Sci,2012,53(48):89-96.
