身材矮小合并46XY性反转1例及文献复习*
2018-04-28何明明李艳英朱惠娟汲宝兰崔维县
何明明 李艳英 朱惠娟 汲宝兰 崔维县 张 梅△
(1济宁医学院附属医院内分泌科,2中国生长发育行为医学研究中心,济宁 272029;3北京协和医院内分泌科,北京 100730)
性反转综合征(sex reversal syndrome,SRS)是一种性别发育异常的人类遗传性疾病,主要表现为性腺性别与染色体性别不相符,其临床分型包括46,XY女性性反转和46,XX男性性反转两类。46,XY 女性性反转综合征又称为 Swyer 综合征,虽然含有决定睾丸发育的Y染色体,但是常缺乏睾丸组织,性腺组织向卵巢方向分化发育,表型为女性,群体发病率约为1/100000[1]。由于性别的决定和分化是一个十分复杂的发育过程,涉及众多基因,在临床表型上具有较大的异质性,因此,诊断较为困难。现报告1例因“身材矮小”就诊的46XY性反患者,并结合文献复习,探讨46,XY性反转综合征的临床特征及诊治思路。
1 病例资料
1.1 患者病史
患者社会性别女性,因“身材矮小3年”于2014年4月13日就诊于济宁医学院附属医院内分泌科。出生日期2000年10月6日,年龄13岁6月,足月剖宫产,出生身长50cm,体重3000g,母亲孕期无特殊用药史。10岁半时发现较同龄儿矮小,12岁曾行左旋多巴生长激素激发试验,30min时生长激素达高峰至6.01ng/ml,应用“重组人生长激素”治疗1月,因效果欠佳停药。近1年身高增长速度为6cm/年,无乳腺发育,无月经来潮,为明确身材矮小原因来诊。既往史:5岁时诊断为“先天性室间隔缺损”并行手术治疗。家族史:父亲身高165cm,母亲身高155cm,父母非近亲结婚,无矮小家族史。
1.2 体格检查
身高139cm(<3rd),体重28Kg,内眦赘皮(+),颈蹼(+),低耳位(+),低发际(+)。女性外观,面部多雀斑,轻度肘外翻,盾状胸。双乳腺未发育(TannerI期),阴毛(TannerI期),幼稚外阴,阴蒂无肥大,大小阴唇无畸形,阴道、尿道分别开口。脊柱、四肢无畸形。
1.3 辅助检查
患者促卵泡刺激素(FSH)、促黄体生成素(LH)水平偏高,雌二醇(E2)水平偏低,皮质醇、促肾上腺皮质激素(ACTH)、甲功、肝肾功、电解质均正常。见表1。

表1 患者实验室检查结果
骨龄片:骨龄10岁(图1)。肾上腺CT平扫:左侧肾上腺内侧支略增粗(图2)。妇科彩超:未见子宫及卵巢组织,双侧腹股沟区可见低回声结节,右侧大小约0.9cm×0.4cm×0.7cm,左侧大小约0.7cm×0.3cm×0.4cm,形态欠规则(图3)。腹股沟探查术,术后病理不支持睾丸组织。两次染色体核型均为46,XY,无嵌合体(图4),Y染色体性别决定区(sex-determining region of Y,SRY)基因检测阳性。

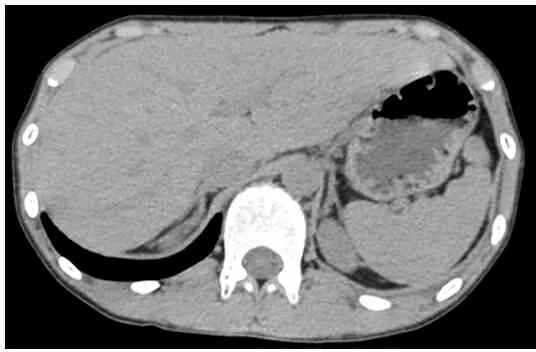
图1 骨龄片
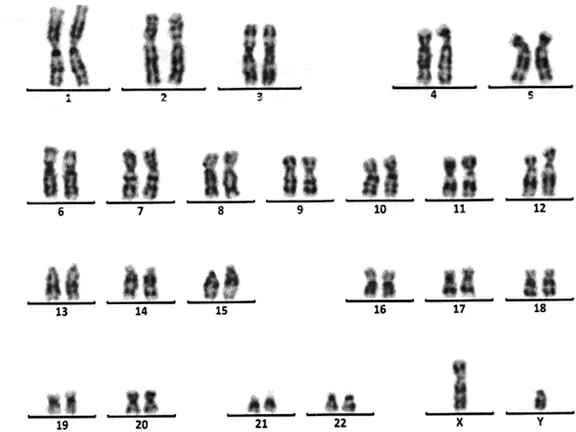
图3 妇科彩超
图4 染色体核型
1.4 诊断、治疗及随访
根据患者临床特征、辅助检查及基因检测结果,诊断为46,XY女性性反转综合征。本例患者社会性别女性,并有女性外阴,患者及家属均选择女性性别,继续按女孩抚养。患者身材矮小,骨龄落后,身高尚有生长空间,但有发生恶性肿瘤的倾向,因此不予重组人生长激素治疗,动态观察身高增长速度。患者年龄16岁9月时,身高增长至148.4cm(<3rd),开始给予“戊酸雌二醇片”1mg/d治疗。规律治疗5月后,身高增长至149.1cm(<3rd),双侧乳腺发育,Tanner分期III期,但外阴仍为幼稚型,阴毛Tanner分期I期。因此,继续该治疗方案,以促进第二性征发育,并定期复查腹部超声,及时发现残余睾丸组织,避免发生恶性肿瘤。
2 讨论
人类性别的分化和决定是一个十分复杂的发育过程,是以SRY基因为主导多种基因有序参与、级联表达的过程。SRY基因位于Y染色体短臂(Yp11.32),其编码产物为睾丸决定因子,可诱导睾丸和男性生殖管道的形成,在男性性腺分化过程中起着调控启动作用[2]。受精卵染色体核型为46,XY时,胚胎发育的第6~8周,原始性腺在SRY基因主导作用下开始出现睾丸分化。同时,SRY基因编码蛋白在胚胎发育第8~10周开始引发苗勒氏管抑制物质(MIS)表达,抑制副中肾管分化为女性内生殖管道,而中肾管在睾酮的作用下可分化为男性内生殖管道,出现男性表型[3]。但SRY基因缺失或者易位时,由于缺乏性别决定基因,原始性腺在胚胎12~13周时可自动分化为卵巢,副中肾管由于缺乏MIS的抑制作用而发育分化成输卵管、子宫,从而出现女性表型,最终发生性反转[4]。近年来研究表明,大约10%~15%的46,XY女性性反转与 SRY 基因突变有关[5],但SRY基因不是决定睾丸发育的唯一要素。Sukumaran等[6-7]的研究显示DAX1(也被称为NROB1)和基因WNT4的复制,或者是SOX9,SF1,WT1及DMRT1-DMRT2等基因的单基因异常,均参与了胚胎中从未分化原始生殖嵴到两性内生殖器官形成的性别决定过程,均与46,XY女性性反转有关。
性分化异常疾病是指在胚胎发育过程中任何阶段出现基因变异或调控因子异常时,表型性别、性腺性别和染色体性别出现不一致的现象。其中,性反转是常见的性别分化异常疾病,根据染色体核型分为46,XX男性性反转和46,XY女性性反转。46,XY 女性性反转综合征患者常因原发性闭经、无女性第二性征或第二性征发育迟缓而就诊[8],在已报道的病例中,均呈女性外表,但生殖器官的发育却各不相同:部分患者具有正常的外生殖器,但无发育正常的性腺;部分患者表现为幼稚型女性外生殖器官,但内生殖器官发育不全;此外还可有正常的外生殖器官,正常的阴道和宫颈,但子宫小于正常[9]。有文献报道特征性的表现为原发性闭经、骨龄落后、成年高身材、第二性征发育和/或外生殖器发育不良[10],但以身材矮小为突出表现者罕见。由于该病发病率较低,在遗传学及临床表现方面具有较大的异质性,常缺乏典型临床表现,临床诊断较困难。
本例患者诊治思路如下:1)身材矮小:患者就诊年龄13岁6月,身高139cm,落后于同龄女童身高的第3百分位,骨龄10岁,矮小症诊断明确。2)性发育延迟:患者13岁6月,无女性第二性征发育,符合性发育延迟。对于矮小症合并第二性征不发育者,LH、FSH水平高,雌二醇、孕酮水平低,应首先考虑Turner综合征。Turner综合征是一种先天性染色体异常所致的疾病。临床表现为身材矮小、生殖器与第二性征不发育及一组躯体发育异常的疾病[11]。染色体异常除典型的45,X外,可有多种嵌合体,如45,X/46,XX、45,X/46,X,i(Xq)、45,X/47,XXX等。本例患者身材矮小,合并先天性心脏病、盾状胸等躯体发育异常,无发育正常的内生殖器官及第二性征,促性腺激素水平高,雌激素水平低,因此首先考虑为Turner综合征。但两次染色体核型均为46,XY,无嵌合体,排除了该病。3)46,XY女性:患者表型性别与染色体性别不一致,为46,XY女性性发育异常,根据2006年欧洲和美国儿科内分泌协会共识,该病根据发病机制分为:1)性腺分化发育异常,主要包括完全性或部分性性腺发育不良(SRY、SOX9、SF1等基因突变)等;2)雄激素合成障碍,主要包括体黄体生成激素(LH)受体突变、雄激素合成各种酶缺乏(17-α羟化酶)等;3)雄激素功能障碍,主要包括雄激素不敏感综合征等[12]。根据本患者临床特征,应与先天性肾上腺皮质增生症中的17-α羟化酶缺陷相鉴别。该酶缺陷时女性(核型46,XX)因雌二醇合成缺乏可引起性幼稚,男性(核型46,XY)因雄激素合成受阻可导致外生殖器呈幼稚女性型,内生殖器为男性型,无腋毛、阴毛生长,青春期后FSH和LH均明显升高,骨龄落后。因糖皮质激素合成受阻,血、尿皮质醇低、血17-羟孕酮低,ACTH高;因盐皮质激素合成途径亢进,表现为高血压、低血钾。该患者无第二性征发育,染色体核型及性激素水平均与该病相符,但无皮质醇及ACTH水平异常,无高血压、低血钾、双侧肾上腺增生等表现,可排除17-α羟化酶缺陷。结合患者临床表现、实验室检查、染色体核型及SRY基因检测阳性,可诊断为46,XY 女性性反转综合征,上述3种疾病的鉴别要点见表2。46,XY性反转综合征患者一般因性腺分化不良而出现性激素水平低下,导致骨骺闭合延迟而出现身材偏高或正常。有个别报道此类患者合并生长激素缺乏而导致身材矮小者[13],该患者以身材矮小就诊,身高<3rd,符合矮小症诊断,行生长激素激发试验峰值6.01ng/ml提示为不完全性生长激素缺乏,考虑患者系矮小症合并46,XY性反转。

表2 主要鉴别疾病临床特征
46,XY性反转综合征的治疗包括手术治疗、药物治疗及心理治疗等,但目标性别选择主要根据生殖器官发育特点、社会性别及本人的意愿等综合考虑。该患者社会性别女性,具有正常女性外阴,患者及家属均愿意继续选择女性性别,因此,给予戊酸雌二醇促进女性第二性征发育。46,XY 女性性反转综合征患者发育不全的性腺无生理功能,但有发生肿瘤的风险,有报道显示20%~30%患者可发生生殖细胞肿瘤,包括性腺母细胞瘤、无性细胞瘤、胚胎癌等[14-15],其发生风险与性分化异常伴Y染色体密切相关[16-17],因此对发育不良的性腺应及时进行手术切除以免恶变。本例患者已行腹股沟探查术,术后病理未见恶变组织,仍需定期复查,及时发现残余性腺组织,避免发生恶性肿瘤。
总结本例诊治体会,以身材矮小就诊的患者,在关注身高生长的同时,还应重视其性发育进展情况。针对性发育延迟患者需进一步探究其病因,慎用重组人生长激素治疗。对于身材矮小、骨龄落后、第二性征发育延迟、内生殖器发育异常、促性腺激素水平高而性激素水平低的患者应进一步行染色体检查,及早完善基因学检测,以建立正确的诊断思路,避免误诊、漏诊,延误治疗。
参考文献:
[1] 蒋三亮,张思仲,杨军.性反转综合征患者的分子病因学研究[J].中华医学遗传学杂志,1993,10(1): 18-20.
[2] 文雪,张元珍.性反转综合征的临床特征及遗传学研究进展[J].中国优生与遗传杂志,2017,25(3): 121-123.
[3] 刘巍,伍学焱,李玉秀,等.第319例女性外阴-斜视-匙状甲-高颚弓-青春期男性化[J].中华医学杂志,2014,94(28):2229-2231.
[4] 武桂芳,金瑞林,贾和平.46,XY女性性反转综合征一例sry基因的错义突变分析[J].解放军医药杂志,2011,23(1): 4-6.
[5] Okuhara K,Tajima T,Nakae J,et al.A novel missense mutation in the HMG box region of the SRY gene in a Japanese patient with an XY sex reversal[J].J Hum Genet,2000,45(2): 112-114.DOI:10.1007/s100380050026.
[6] Sukumaran A,Desmangles JC,Gartner LA,et al.Duplication of dosage sensitive sex reversal area in a 46,XY patient with normal sex determining region of Y causing complete sex reversal[J].J Pediatr Endocrinol Metab,2013,26(7-8): 775-779.DOI:10.1515/jpem-2012-0354.
[7] Domenice S,Corrêa RV,Costa EM,et al.Mutations in the SRY,DAX1,SF1 and WNT4 genes in Brazilian sex-reversed patients[J].Revista brasileira de pesquisas medicas e biologicas,2004,37(1): 145-150.DOI:10.1590/s0100-879x2004000100020.
[8] Ben Temime R,Chachial A,Attial L,et al.46 XY pure gonadal dysgenesis with gonadoblastoma and dysgerminoma[J].Tunis Med,2008,86(7): 710-713.
[9] Michala L,Goswami D,Creighton SM,et al.Swyer syndrome: presentation and outcomes[J].BJOG,2008,115(6): 737-741.DOI:10.1111/j.1471-0528.2008.01703.x.
[10] DU X,Zhang X,Li Y,et al.46,XY female sex reversal syndrome with bilateral gonadoblastoma and dysgerminoma[J].Exp Ther Med,2014,8(4): 1102-1104.DOI:10.3892/etm.2014.1922.
[11] Saenger P,Wikland KA,Conway GS,et al.Recommendations for the diagnosis and management of Turner syndrome[J].J Clin Endocrinol Metab,2001,86(7): 3061-3069.DOI:10.1210/jcem.86.7.7683.
[12] Hughes IA,Nihoul-Fékété C,Thomas B,et al.Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development[J].Best Pract Res Clin Endocrinol Metab,2007,21(3): 351-365.DOI:10.1016/j.beem.2007.06.003.
[13] 金文胜,李佳,李红梅,等.Swyer综合征合并生长迟缓1例[J].广东医学,2014,35(17): 2784.
[14] 曹泽毅.中华妇产科学[M].2 版.北京:人民卫生出版社,2005.2689.
[15] Hersmus R,Stoop H,Turbitt E,et al.SRY mutation analysis by next generation (deep) sequencing in a cohort of chromosomal Disorders of Sex Development (DSD) patients with a mosaic karyotype[J].BMC Med Genet,2012,13: 108.DOI:10.1186/1471-2350-13-108.
[16] Gibbons B,Tan SY,Yu CC,et al.Risk of gonadoblastoma in female patients with Y chromosome abnormalities and dysgenetic gonads[J].J Paediatr Child Health,1999,35(2): 210-213.DOI:10.1046/j.1440-1754.1999.00319.x.
[17] Fernandes GC,Sathe PA,Naik LP,et al.Bilateral gonadoblastomas with unilateral dysgerminoma in a case of 46 XY pure gonadal dysgenesis (Swyer syndrome)[J].Indian J Pathol Microbiol,2010,53(2): 376-378.DOI:10.4103/0377-4929.64292.
