PPAR-γ通过活化PTEN抑制肾间质成纤维细胞MMP2的活化
2018-04-24卢家美张苏梅杨艳艳吕治安付荣国
卢家美,刘 丹,张苏梅,杨艳艳,吕治安,韩 锦,付荣国
(1. 西安交通大学第二附属医院肾病内科,陕西西安 710004;2. 西安医学院护理学院,陕西西安 710021)
肾间质纤维化(renal interstitial fibrosis, RIF)是各种肾脏疾病进展为终末期肾病(end-stage renal disease, ESRD)的共同途径和主要病理学改变[1-2],其发病机制为各种细胞因子及炎症介质诱导的肾成纤维细胞等间质细胞增殖及细胞外基质(extracellular matrix, ECM)的异常沉积。而基质金属蛋白酶(matrix metalloproteinases, MMPs)与基质金属蛋白酶组织抑制剂 (tissue inhibitor of metalloproteinases, TIMPs)的失衡被认为是上述病理改变的主要分子机制[3]。其中,MMP家族的异常活化被证实在上述病理生理过程中起关键作用。MMP2作为MMP家族主要成员,其异常活化在肾间质纤维化中发挥尤其重要的作用。MMP2作为一种降解胶原的主要蛋白水解酶,主要由肾间质成纤维细胞以水溶性酶原的形式分泌至细胞外,其在蛋白裂解酶的作用下,通过脱去部分前肽转变为有活性的形式,从而发挥其相应的分子生物学作用[4]。因此,探索肾成纤维细胞MMP2活化的分子机制并寻求有效的治疗靶点对预防及治疗肾间质纤维化有着十分重要的意义。
血小板源性生长因子(platelet-derived growth factor, PDGF)是一种重要的促细胞分裂剂,可刺激多种细胞的分裂与增殖,广泛参与多个组织器官的生理活动和病理损伤过程[5]。其过度表达在肾间质纤维化中的作用日益受到重视。进一步研究发现,PDGF主要通过诱导肾成纤维细胞活化增殖及细胞外基质异常沉积参与肾间质纤维化的发生[6]。但是,PDGF通过何种信号机制及分子介质发挥上述分子生物学作用目前尚不完全明确。MMP2作为肾间质纤维化的关键分子介质,被证实广泛参与肾间质纤维化的发病。研究还发现,磷脂酰肌醇-3-激酶/蛋白激酶B(P13K/AKT)介导了PDGF诱导的肾间质纤维化的发生[7]。本研究将探讨PDGF是否通过激活P13K/AKT信号通路活化MMP2从而诱导肾间质纤维化。
过氧化物酶体增殖物激活受体-γ(peroxisome proliferators activated receptor-γ, PPAR-γ)作为一种重要的核转录因子,隶属于Ⅱ型核受体超家族成员。进一步研究发现,罗格列酮通过激活PPAR-γ广泛抑制多种细胞的增殖、诱导凋亡及分化[8]。研究还发现,PPAR-γ激动剂通过活化人磷酸酶及张力蛋白同源的基因(phosphatase and tensin homolog deleted on chromosome ten, PTEN)特异性阻抑AKT信号通路,进而抑制肺成纤维细胞的活化、增殖[9-10]。本研究将探讨激活的PPAR-γ是否通过活化PTEN抑制PI3K/AKT信号通路降低MMP2活性,进而抑制肾间质纤维化的发生。
1 材料与方法
1.1原代肾间质成纤维细胞的分离及培养取4~6周龄雌性Balb/c小鼠,颈椎脱臼法处死。采用FREDRIKSSON等[11]方法获取小鼠原代肾间质成纤维细胞。无菌条件下分离并获取肾髓质,将肾髓质剪成细小的组织块,置于含0.5 mg/mL弹性蛋白酶(Sigma)、1.5 mg/mL胶原酶(Sigma)的溶液中培养30 min,离心收集细胞并培养于含150 mL/L胎牛血清、100 U/mL青霉素和100 μg/mL链霉素的DMEM高糖培养基中。以1.25 g/L胰酶消化细胞并传代。所培养的细胞经异硫氰酸荧光素(FITC)标记的波形蛋白(Sigma)染色,证实成纤维细胞阳性率大于95%。下述所有实验均在4~8代细胞中进行,干预前均预先给予10 mL/L胎牛血清饥饿12 h。以PDGF-AA(Sigma)刺激原代培养的肾成纤维细胞诱导其增殖,LY294002(Sigma)抑制PI3K/AKT信号通路,GW9662(Sigma)、bpV(Pic;Sigma)分别抑制PPAR-γ、PTEN活性,罗格列酮购自Alexis公司。
1.2明胶酶谱检测MMPs活性将含等量总蛋白的培养基上清上样于含10 g/L明胶的胶中,以90、120 V恒压电泳,切取包含72、92 ku大小的凝胶。置于洗脱液中震荡洗脱2次(20 min/次),漂洗液震荡漂洗2次(20 min/次)。将凝胶置于孵育液中37 ℃孵育48 h;考马斯亮蓝染色2 h,依次脱色后1.75 mol/L醋酸显色。
1.3免疫印迹法检测蛋白水平以RIPA裂解液(Bioteke Corporation)破碎原代培养的肾间质成纤维细胞,4 ℃下12 000 r/min离心10 min,收集裂解液上清以获取细胞总蛋白。以BCA试剂盒(Pierce)检测细胞总蛋白浓度,等量上样于100 g/L SDS-PAGE胶,采用半干转膜法将蛋白质转移至硝酸纤维素膜(NC;Bio-Rad),50 g/L脱脂奶粉封闭2 h。以抗-p-AKT(Cell signal technology)、t-AKT(Cell signal technology)、TIMP1/TIMP2/TIMP3(Cell signal technology)、PPAR-γ(Cell signal technology)和GAPDH(Sigma)的兔源性抗体作为一抗,以辣根过氧化物酶(HRP)标记的羊抗兔IgG(Sigma)作为二抗,West Pico化学荧光底物(Pierce)激发荧光并曝光于感光胶片,采用Scion NIH图像分析软件检测免疫印迹强度,以检测细胞中上述蛋白的表达水平。
1.4PTEN活性检测采用PTEN 活性检测ELISA试剂盒检测PTEN的活性(Echelon biosciences Inc),操作方法按照说明书进行。获取细胞裂解液上清,每孔100 μL等量上样,37 ℃孵育2 h,TBST洗板3次,每次20 min,于450 nm波段下读取A450值。
1.5统计学处理实验数据以均数±标准差表示,所有实验数据均符合正态性和方差齐性。采用SPSS 13.0进行数据统计与分析,采用单因素方差分析(One-way ANOVA)进行组间数据的比较,Tukey post hoc检验进行组间的多重比较。以P<0.05为差异有统计学意义。
2 结 果
2.1PDGF对肾间质成纤维细胞MMPs/TIMPs活性的刺激作用基质金属蛋白酶(MMPs)/基质金属蛋白酶组织抑制剂(TIMPs)的失衡被证实是细胞外基质(ECM)异常沉积乃至肾间质纤维化的重要发病基础,为明确PDGF通过上述何种分子介质发挥诱导肾间质纤维化的作用,采用不同浓度的PDGF-AA(1、3、10、30 ng/mL)刺激肾间质成纤维细胞24 h,对照组以与其他组等体积的PDGF-AA溶媒DMSO刺激细胞。采用明胶酶谱法检测MMP2、MMP9的活性。结果显示PDGF-AA剂量依赖性活化MMP2,峰浓度为10 ng/mL,而对MMP9无此作用(图1A)。提取细胞裂解液,采用免疫印迹法检测TIMP1、TIMP2、TIMP3水平。结果发现,PDGF-AA仅引起TIMP1的轻度升高,但其差异无统计学意义(P=0.179,P>0.05,与对照组相比),而对TIMP2、TIMP3的表达没有影响(图1B)。
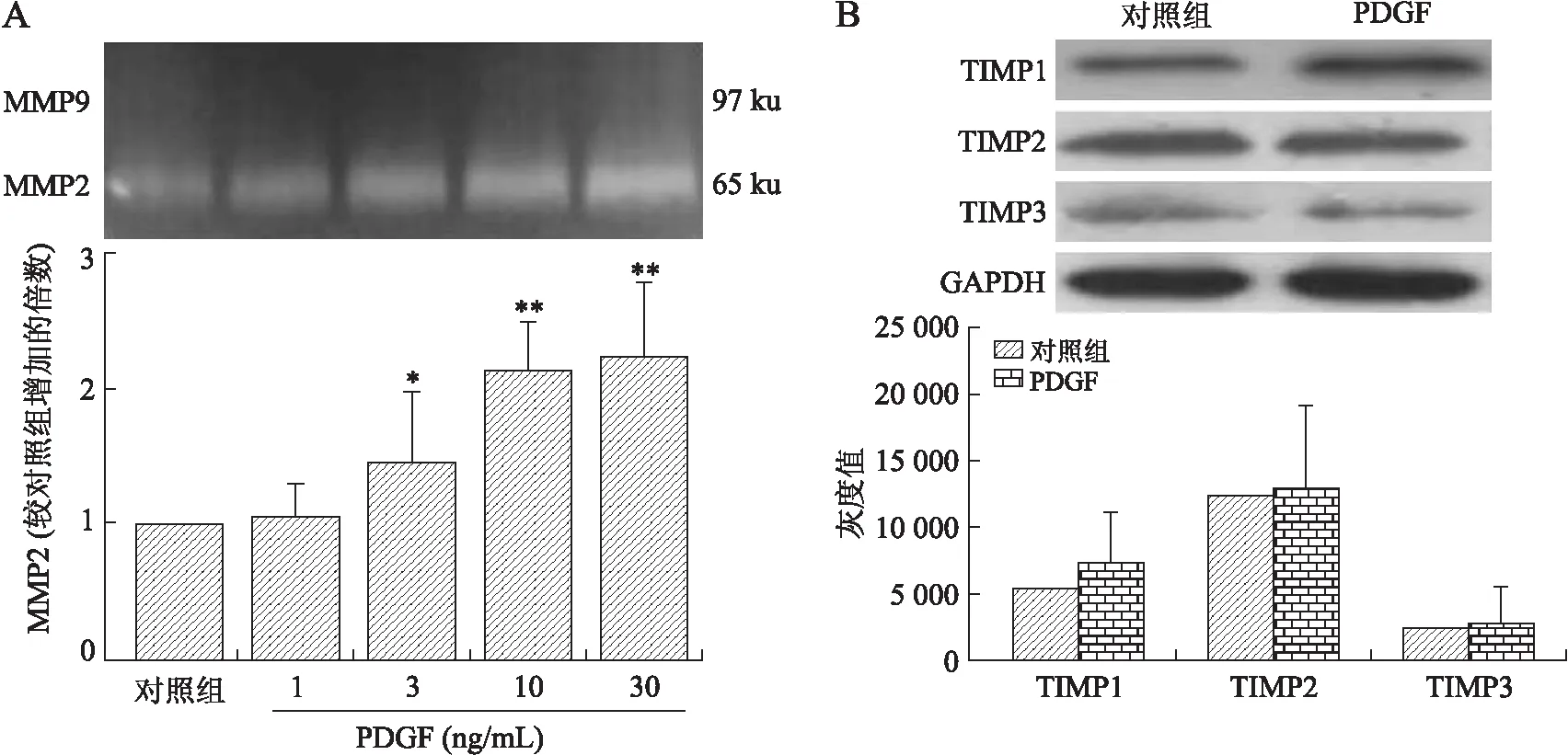
图1PDGF对肾间质成纤维细胞MMPs/TIMPs活性的作用
Fig.1 Effects of PDGF on MMPs/TIMPs activation or production in mouse renal interstitial fibroblasts (n=3 in each group)
与对照组相比,*P<0.05,**P<0.01。
2.2AKT介导PDGF诱导的肾间质成纤维细胞MMP2活化为探讨PDGF诱导肾间质成纤维细胞MMP2活化的分子机制,10 ng/mL PDGF-AA刺激细胞30 min,采用免疫印迹法检测p-AKT的水平。结果发现,PDGF-AA显著诱导AKT的磷酸化活化(P=0.000 1,P<0.01,与对照组相比;图2A)。为进一步明确激活的PI3K/AKT信号通路是否特异性介导了MMP2的活化,于PDGF-AA刺激细胞前1 h,预先予以PI3K抑制剂LY294002(50 μmol/L)孵育细胞,PDGF-AA干预细胞后24 h,检测MMP2活性。结果发现,抑制PI3K/AKT信号通路显著抑制PDGF-AA诱导的MMP2活化(P=0.002 3,P<0.01,与PDGF组相比;图2B)。
2.3PPAR-γ抑制PDGF诱导的肾间质成纤维细胞MMP2活化激活的PPAR-γ被证实显著抑制慢性肾脏病动物模型肾间质纤维化的发生,然而其分子机制尚不明确[9]。本研究旨在探讨PPAR-γ是否通过抑制PDGF诱导的MMP2活化抑制肾间质纤维化。首先,采用PPAR-γ激动剂罗格列酮(10 μmol/L)刺激肾间质成纤维细胞,发现罗格列酮可显著激活PPAR-γ(P=0.004 2,P<0.01,与对照组相比;图3A)。预先予以PPAR-γ抑制剂GW9662(10 μmol/L)预处理细胞,可显著逆转罗格列酮对PPAR-γ的激活(P=0.001 2,P<0.01,与罗格列酮组相比;图3A)。进一步,于PDGF-AA刺激肾间质成纤维细胞前1 h,采用PPAR-γ激动剂罗格列酮(10 μmol/L)刺激细胞,发现激活的PPAR-γ显著抑制PDGF-AA诱导的MMP2活化(P=0.000 1,P<0.01,与PDGF组相比;图3B)。采用GW9662特异性阻抑PPAR-γ的活性,可显著逆转罗格列酮对MMP2活性的抑制作用(P=0.000 1,P<0.01,与PDGF+Rosi组相比;图3B)。

图2AKT介导PDGF诱导的肾间质成纤维细胞MMP2活化
Fig.2 AKT mediated PDGF-induced MMP2 activation in mouse renal interstitial fibroblasts (n=3 in each group)
与对照组相比,**P<0.01;与PDGF组相比,# #P<0.01(LY29:LY294002)。

图3PPAR-γ抑制PDGF诱导的肾间质成纤维细胞MMP2活化
Fig.3 Rosiglitazone (Rosi) inhibited PDGF-stimuated MMP2 activation in renal interstitial fibroblasts (n=3 in each group)
与对照组相比,**P<0.01;与PDGF组相比,##P<0.01;与Rosi PDGF组相比,△△P<0.01(Rosi:罗格列酮组)。
2.4PPAR-γ通过激活PTEN抑制AKT磷酸化以抑制PDGF诱导的肾间质成纤维细胞MMP2活化为进一步明确激活的PPAR-γ抑制PDGF诱导的肾间质成纤维细胞MMP2活化的分子机制,分别于PDGF-AA刺激细胞前1 h予以10 μmol/L的罗格列酮预处理细胞,PDGF-AA干预后半小时提取细胞裂解液上清,采用PTEN活性ELISA检测试剂盒测定PTEN活性。发现激活的PPAR-γ显著活化PTEN(P=0.000 0,P<0.01,与PDGF组相比;图4A),而特异性阻抑PPAR-γ活性后,PTEN的活性显著降低(P=0.001 5,P<0.01,与Rosi PDGF组相比;图4A),而PDGF-AA对PTEN的活性没有影响(P=0.467 1,P>0.05,与对照组相比;图4A)。进一步,于PDGF-AA刺激细胞前1 h予以罗格列酮或同时予以罗格列酮+10 μmol/L PTEN抑制剂bpV(pic; Cayman, Ann Arbor)刺激细胞,以明确激活的PPAR-γ是否通过活化PTEN抑制AKT磷酸化发挥抑制MMP2的作用。结果发现,抑制PTEN可逆转PPAR-γ激活对AKT磷酸化、MMP2活化的抑制作用(P=0.003 9,P<0.01,与Rosi PDGF组相比;图4B、4C)。提示PPAR-γ通过激活PTEN抑制PDGF诱导的肾间质成纤维细胞AKT磷酸化进而抑制MMP2活化。

图4PPAR-γ通过激活PTEN抑制AKT磷酸化以抑制PDGF诱导的肾间质成纤维细胞MMP2活化
Fig.4 PPAR-γ suppressed PDGF-induced MMP2 generation by activation of PTEN and resultant inhibition of AKT phosphorylation (n=3 in each group)
与对照组相比,**P<0.01;与PDGF组相比,##P<0.01;与PDGF+Rosi组相比,△△P<0.01(Rosi:罗格列酮;LY29:LY294002;Pic:PTEN抑制剂)。
3 讨 论
本研究揭示PDGF通过激活肾间质成纤维细胞PI3K/AKT信号通路活化MMP2。而PDGF的上述作用可被激活的PPAR-γ抑制。进一步研究发现,PPAR-γ的上述作用通过激活PTEN实现[12]。本研究提示,激活PPAR-γ有望成为慢性肾脏病肾间质纤维化的新的治疗靶点。
基质金属蛋白酶(MMPs)/基质金属蛋白酶组织抑制剂(TIMPs)的平衡被证实在维持细胞外基质生成及降解过程中发挥重要作用[13]。在慢性肾脏病患者及动物模型中均证实MMPs/TIMPs失衡广泛参与肾间质纤维化的发病[14]。而在MMP的众多亚型中,MMP2被证实与肾脏重塑的关系最为密切。MMP2通过降解非纤维结构样胶原并裂解弹性蛋白以释放具有生物活性的ECM碎片,其破坏弹性蛋白间的致密结构,使肾间质成纤维细胞迁移至上皮下,并在一系列刺激因子的作用下,肾间质成纤维细胞被活化、增殖并分泌细胞外基质,从而启动肾间质纤维化的发生[14-17]。
PDGF作为成纤维细胞及平滑肌细胞等多种间充质细胞的促有丝分裂原及集落刺激因子,被证实除刺激细胞增殖外,还具有诱导上述细胞分泌细胞外基质的作用[6]。而PDGF的上述作用是其在肾间质纤维化发病中发挥重要作用的分子生物学基础。因此,针对PDGF引起的发病机制的治疗,将成为慢性肾脏病肾间质纤维化治疗的重要靶点。本研究发现,PDGF剂量依赖性活化MMP2,而MMP9等其他亚型在肾间质成纤维细胞中活性较低[18-19]。PDGF对TIMP的表达的影响亦不明显。进一步研究发现,PDGF诱导MMP2活化的作用是通过激活PI3K/AKT信号通路实现的。本研究提示PI3K/AKT信号通路介导的MMPs/TIMPs失衡与肾间质纤维化密切相关。
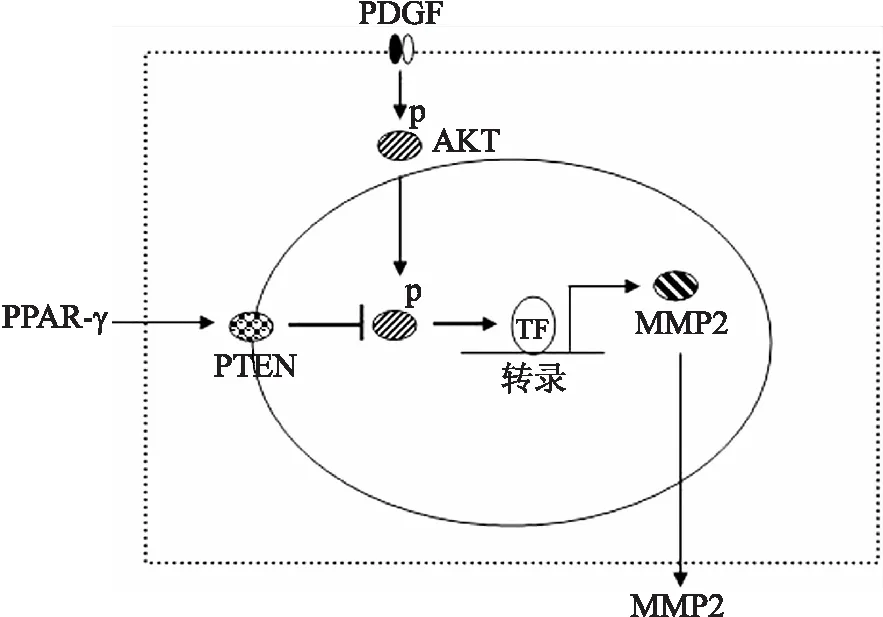
图5PPAR-γ抑制PDGF诱导的肾间质成纤维细胞MMP2活化的分子机制
Fig.5 Proposed mechanisms of the inhibition of PPAR-γ on PDGF-induced MMP2 activation in renal interstitial fibroblasts
PPAR-γ作为重要的内源性核转录因子,被证实可通过抑制肾间质成纤维细胞活化、增殖及细胞外基质异常沉积抑制慢性肾脏病肾间质纤维化的发生[9,20]。研究发现,MMP2介导了慢性肾脏病动物模型肾间质纤维化的发生[14,21]。然而,激活的PPAR-γ是否通过抑制MMP2活化发挥上述作用目前尚不明确。本研究发现,激活的PPAR-γ显著抑制PDGF诱导的MMP2活化;而PPAR-γ的上述作用是通过激活PTEN从而抑制AKT磷酸化实现的。PTEN作为一种重要的抑癌基因,还发挥调控细胞生长、增殖、迁移及能量代谢的作用,而PTEN的上述分子生物学作用主要通过抑制PI3K/AKT信号通路实现(图5)。
目前,多种PPAR-γ激动剂已经广泛用于临床治疗糖尿病,一系列动物实验也证实激活的PPAR-γ对肿瘤、肺间质纤维化、慢性支气管炎、冠心病等具有显著的保护作用[22]。本研究结果提示,激活的PPAR-γ具有潜在抑制肾间质纤维化的作用,为慢性肾脏病肾间质纤维化的治疗提供了新的治疗方法。然而,PPAR-γ激动剂能否用于慢性肾脏病肾间质纤维化的治疗,尚需一系列的研究以证实其安全性及有效性。
参考文献:
[1] EDDY AA. Molecular basis of renal fibrosis[J]. Pediatr Nephrol, 2000, 15(3):290-301.
[2] CHUANG PY, MENON MC, HE JC. Molecular targets for treatment of kidney fibrosis[J]. J Mol Med (Berl), 2013, 91(5):549-559.
[3] TAN RJ, LIU Y. Matrix metalloproteinases in kidney homeostasis and diseases[J]. Am J Physiol Renal Physiol, 2012, 302(5):1351-1361.
[4] ZHAO H, DONG Y, TIAN X, et al. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases[J]. World J Nephrol, 2013, 2(8):84-89.
[5] FLOEGE J, EITNER F, ALPERS CE. A new look at platelet-derived growth factor in renal disease[J]. J Am Soc Nephrol, 2008, 19(1):12-23.
[6] ROEYEN CR, OSTENDORF T, FLOEGE J. The platelet-derived growth factor system in renal disease: An emerging role of endogenous inhibitors[J]. Eur J Cell Biol, 2012, 91(3):542-551.
[7] ZVIBEL I, BAR-ZOHAR D, KLOOG Y, et al. The effect of Ras inhibition on the proliferation, apoptosis and matrix metalloproteases activity in rat hepatic stellate cells[J]. Dig Dis Sci, 2008, 53(7):1048-1053.
[8] YESSOUFOU A, WAHLI W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels[J]. Swiss Med Wkly, 2010, 140(5):13071-13074.
[9] KAWAI T, MASAKI T, DOI S, et al. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β[J]. Lab Invest, 2009, 89(1):47-58.
[10] BOOR P, OSTENDORF T, FLOEGE J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets[J]. Nat Rev Nephrol, 2010, 6(11):643-656.
[11] FREDRIKSSON L, LI H, FIEBER C, et al. Tissue plasminogen activator is a potent activator of PDGF-CC[J]. EMBO J, 2004, 23(19):3793-3802.
[12] WANG L, ZHAO Y, GUI B, et al. Acute stimulation of glucagon secretion by linoleic acid results from GPR40 activation and [Ca2+]i increase in pancreatic islet {alpha}-cells[J]. J Endocrinol, 2011, 210(2):173-179.
[13] CONSOLO M, AMOROSO A, SPANDIDOS DA, et al. Matrix metalloproteinases and their inhibitors as markers of inflammation and fibrosis in chronic liver disease (Review)[J]. Int J Mol Med, 2009, 24(4):143-152.
[14] DU X, SHIMIZU A, MASUDA Y, et al. Involvement of matrix metalloproteinase-2 in the development of renal interstitial fibrosis in mouse obstructive nephropathy[J]. Lab Invest, 2012, 92(3):1149-1160.
[15] DUYMELINCK C, DENG JT, DAUWE SE, et al. Inhibition of the matrix metalloproteinase system in a rat model of chronic cyclosporine nephropathy[J]. Kidney Int, 1998, 54(5):804-818.
[16] CAI G, ZHANG X, HONG Q, et al. Tissue inhibitor of metalloproteinase-1 exacerbated renal interstitial fibrosis through enhancing inflammation[J]. Nephrol Dial Transpl, 2008, 23(9):1861-1875.
[17] WANG X, ZHOU Y, TAN R, et al. Mice lacking the matrix metalloproteinase-9 gene reduce renal interstitial fibrosis in obstructive nephropathy[J]. Am J Physiol Renal Physiol, 2010, 299(7):973-982.
[18] CHEN YT, CHANG FC, WU CF, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis[J]. Kidney Int, 2011, 80(11):1170-1181.
[19] BOOR P, FLOEGE J. Chronic kidney disease growth factors in renal fibrosis[J]. Clin Exper Pharmacol Physiol, 2011, 38(7):441-450.
[20] KULKARNI AA, THATCHER TH, OLSEN KC, et al. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: Implications for therapy of fibrosis[J]. PLoS One, 2011, 6(5):15909-15912.
[21] CHENG S, LOVETT DH. Gelatinase A(MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation[J]. Am J Pathol, 2003, 162(6):1937-1949.
[22] BOLEN S, FELDMAN L, VASSY J, et al. Systematic review: Comparative effectiveness and safety of oral medications for type 2 diabetes mellitus[J]. Ann.Intern Med, 147(2):386-399.
