高效液相色谱-质谱/质谱联用法测定鸡蛋中氯霉素残留量的不确定度评定
2018-04-19黄伟蓉张朋杰张宪臣陈丽斯卢俊文陈润雯
◎ 黄伟蓉,张朋杰,张宪臣,陈丽斯,杨 蕙,卢俊文,陈润雯
(中山海关技术中心,广东 中山 528400)
氯霉素是一种广谱性抗生素,对革兰氏阴性菌和阳性菌均有良好的抑制作用,广泛应用于动物细菌感染的临床治疗;但也能引起人的再生障碍性贫血、粒状白细胞缺失症、新生儿灰色综合症等[1-2];因此食品中氯霉素的残留会对人体健康造成严重危害。中华人民共和国农业部公告第235 号《动物性食品中兽药最高残留限量》将氯霉素列入禁用药,其检测限为 0.1 μg·kg-1。我国规定在鸡肉中的残留限量为100 μg·kg-1,这一限度不会引起骨髓抑制或再生障碍性贫血,但对胚胎有毒性,妊娠动物禁用,产蛋鸡禁用,在鸡蛋中不得检出[3]。鸡蛋也是动物源性食品的重要部分,因此准确测定鸡蛋中氯霉素的残留量具有重要意义。而不确定度的大小直接反应了测量水平的高低,对质量控制具有重要意义;因此为了检测检测结果的可信度,本研究参考了GB/T 22338-2008《动物源性食品中氯霉素类药物残留量测定》中的高效液相色谱-质谱/质谱法检测鸡蛋中的氯霉素残留量,并根据CNAS-GL 06-2006《化学分析中不确定度的评估指南》[4],JJF 1059.1-2012《测量不确定度与表示》[5]及JJG 196-2006《常用玻璃量器检定规程》[6]等相关指导性文件,建立了一套合理、完整的鸡蛋中氯霉素残留量的不确定度评估方案[7-11]。
1 仪器与试剂
AB Sciex Triple Quad 5500 液相色谱串联质谱:配电喷雾离子源(ESI)(美国AB 公司);天平 HF-2 000,感量0.01 g;Sartorius BSA2245,感量0.000 1 g。 搅拌机、振荡器、离心机、氮吹仪、移液器(100 μL、200 μL、100 μL)以及100 mL 容量瓶,A 级。氯霉素标准品,纯度≥99.24%;氘代氯霉素内标,纯度≥99.9%。乙酸乙酯,甲醇,正己烷:色谱纯;无水硫酸钠、氨水:分析纯;0.22μm 有机相滤膜;实验室一级水。
2 方法
2.1 样品的选择和制备
从市场购买1 kg 鲜鸡蛋作为试样。取鲜鸡蛋约500 g,去壳经高速组织搅拌机均匀捣碎混匀,装入洁净容器,密封,待测定。
2.2 标准溶液的配制
浓度为100 μg·mL-1的氯霉素标准储备液:按纯度折算准确称取氯霉素标准物质0.010 0 g,置于100 mL容量瓶中,用甲醇超声溶解稀释并定容至刻度,摇匀。
浓度为1 μg·mL-1的氯霉素标准中间液:用移液器准确移取浓度为100 μg·mL-1的标准储备液1 mL 至100 mL 容量瓶中,用甲醇定容至刻度。
浓度为5 ng·mL-1的氯霉素标准工作液:用移液器准确移取浓度为1 μg·mL-1的标准中间液0.5 mL 至100 mL 容量瓶中,用甲醇定容至刻度。
2.3 样品前处理
准确称取搅拌均匀的鸡蛋样品5 g(精准至0.01 g)置于50 mL 离心管,依次加入15 mL 乙酸乙酯,0.5 mL 氢氧化铵,5 g 无水硫酸钠,振摇10 min,4 500 r·min-1离心10 min,吸取上清液到另一50 mL 离心管中。另取15 mL 乙酸乙酯和0.5 mL 氢氧化铵溶解残渣,重复提取一次。将两次提取液合并,并涡旋混匀。取6 mL提取液于氮吹管中在50 ℃下吹至近干,加2 mL 水溶解,溶解后加3 mL10%甲醇饱和正己烷涡旋混合, 4 500 r·min-1离心5 min,弃去上清液,用10%甲醇饱和正己烷重复净化一次。取下层过0.2 μm 滤膜供高效液相色谱-质谱串联仪。
3 仪器条件
3.1 色谱条件
色谱柱:C18柱(2.1 mm×100 mm,3.0 µm);流动 相:甲醇(A)-水(B),梯度洗脱条件:0 ~2 min, 25%A;2 ~4 min,80%A;4 ~6 min,25%A; 流 速0.4 mL·min-1;柱温:35 ℃;进样量:10 µL。
3.2 质谱条件
电喷雾离子源(ESI);负离子模式采集;喷雾电 压:-4 500 V;气帘气流速:30 L·min-1;锥孔气流速:45 L·min-1;去溶剂气流速:50 L·min-1;多反应监测扫描模式(MRM)。
4 建立不确定度数学模型
氯霉素不确定度数学模型为:

式(1)中:X-试样中氯霉素的残留量,μg·kg-1; c-由标准曲线得出样品中氯霉素的浓度,ng·mL-1;V-试样的最终定容体积,mL;m-样品称样量,g;R-回收率校正因子;fr-LC-MS/MS 仪器的校准因子。
5 不确定度分量的主要来源及其分析
从测量过程和数学模型分析,测量不确定度的主要来源为:标准物质纯度引入的不确定度,标准物质称量引入的不确定度,标准物质配制引入的不确定度,试样称量引入的不确定度,试样最终定容体积引入的不确定度,试样重复测定引入的不确定度,拟合标准曲线引入的不确定度,LC-MS/MS 仪器定量校准引入的不确定度,样品提取净化过程引入的不确定度(以回收率表示)。
针对不同的不确定度来源进行A 类与B 类评定分析。根据实验过程中随机分布和接近正态分布的情况,重复测量试样并获得一组测量值及标准差和使用检定证书、校正证书及仪器的准确度等级等数据作为测量不确定度的分量,利用贝赛尔法进行分析并求出标准不确定度和标准相对不确定度,进而求出合成不确定度和扩展不确定度。
5.1 氯霉素标准物质纯度引入的不确定度
根据B 类评定,标准物质证书给出的纯度为99.24%±0.77%,按照均匀分布计算,标准物质纯度引入的相对不确定度为:

5.2 氯霉素标准物质称量引入的不确定度
根据B 类评定,天平Sartorius BSA2245 的检定证 书,感量为0.000 1 g,称取0.010 0 g 氯霉素标准物质,按照均匀分布计算,其相对不确定度为:

5.3 标准物质配制引入的不确定度
5.3.1 配制氯霉素标准储备液产生的不确定度
准确称取氯霉素标准物质0.010 0 g,置于100 mL的容量瓶中,用甲醇超声溶解稀释并定容至刻度。根据JJG-196-2006《常用玻璃器具检定规程》,A 级 100 mL 容量瓶的最大允许误差为±0.10 mL,按均匀分布计算,其相对不确定度为:

实验室温度为(20±5)℃,甲醇的膨胀系数为1.18×10-3℃-1,以20 ℃时1 mL 体积作为校准,按均匀分布计算,由温度引入的相对不确定度为:

因此配制浓度为100 μg·mL-1氯霉素的标准储备液所引入的合成相对不确定度为:

5.3.2 配制氯霉素标准中间液产生的不确定度
配制过程中使用到100 mL 容量瓶和1 000 μL 的单道移液器。
使用量程为1 000 μL 的单道移液器移取1 mL 时的精度等级为±0.6%(仪器商提供),假设均匀分布,移液器引入的相对不确定度为:

实验室温度为(20±5)℃,甲醇的膨胀系数为1.18×10-3℃-1,温度引入的相对不确定度为:
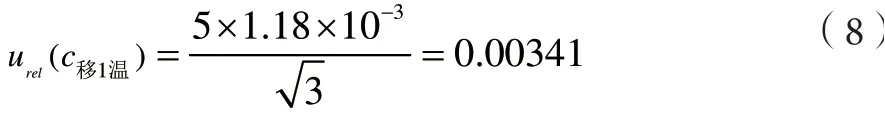
移液器吸取1 mL 引入的相对标准不确定度为

JJG-196-2006《常用玻璃器具检定规程》规定,A级100 mL 容量瓶的最大允许误差为±0.10 mL,按均匀分布计算,其相对不确定度为:

实验室温度为(20±5)℃,甲醇的膨胀系数为1.18×10-3℃-1,以20 ℃时1 mL 体积作为校准,按均匀分布计算,由温度引入的相对不确定度为:

因此配制浓度为1 μg·mL-1氯霉素的标准中间液所引入的合成相对不确定度为:

所以1μg·mL-1氯霉素标准中间液配制过程中所引入的合成相对不确定度为:

5.3.3 配制氯霉素标准工作液引入的不确定度
配制过程中使用到100 mL 容量瓶和1 000 μL 的单道移液器。
使用量程为1 000μL 的单道移液器移取0.5 mL 时的精度等级为±0.8%(仪器商提供),假设均匀分布,移液器引入的不确定度为:

其他指标与5.3.2 一致,移液器吸取0.5 mL 引入的相对标准不确定度为:

根据JJG-196-2006《常用玻璃器具检定规程》,A级100 mL 容量瓶的最大允许误差为±0.10mL,按均匀分布计算,其相对不确定度为:
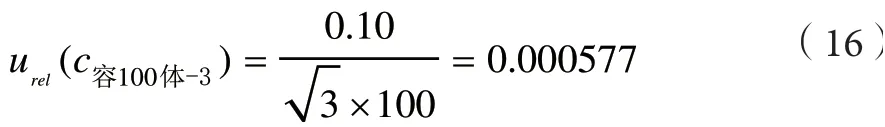
其他指标与5.3.2 中一致,由温度引入的相对不确定度为:

因此配制浓度为5 ng·mL-1的氯霉素标准工作液所引入的合成相对不确定度为:

所以浓度5ng·mL-1氯霉素标准工作液配制过程中所引入的合成相对不确定度为:
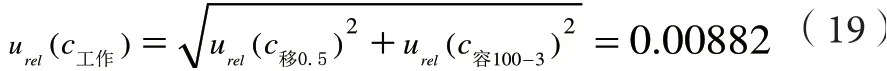
综上所述,所有标准物质溶液配制过程中所引入的合成相对不确定度为:

5.4 试样称量引入的不确定度
鸡蛋样品称样量为5.00 g,所用天平为HF-2000,在0 ~500 g 称量范围内的最大允许误差为±0.01 g,按均匀分布评定,引入的标准不确定度为:

5.5 试样最终定容体积引入的不确定度
使用量程为5 mL 的移液器移取2 mL 水溶解残渣,精度等级为±0.8%(仪器商提供),假设均匀分布,移液器引入的相对不确定度为:

水的体积膨胀系数为2.1×10-4℃-1,以20 ℃时1 mL 体积作为校准,试验温度变化范围为(20±5)℃,按均匀分布计算,由温度引入的相对不确定度为:

所以,样品定容体积产生的相对不确定度为:

5.6 试样重复测定所引入的不确定度
在相同的条件下对样品重复测定6 次,测量结果见表1。
样品测量结果的标准偏差为(共测量6 次,p=6):

表1 样品中氯霉素残留测定结果表
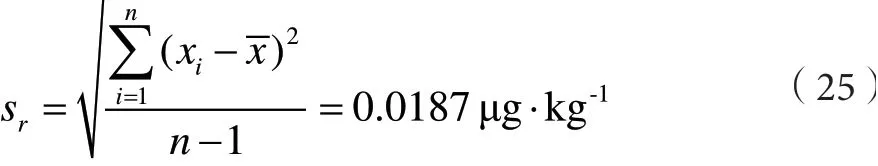
则A 类标准不确定度为:

因此重复测量样品引入的相对不确定度为:

5.7 拟合标准曲线引入的不确定度
拟合引入的标准不确定度计算公式如式(28):

式(28)中:sb为标准溶液响应信号残差的标准差,计算公式如式(29):
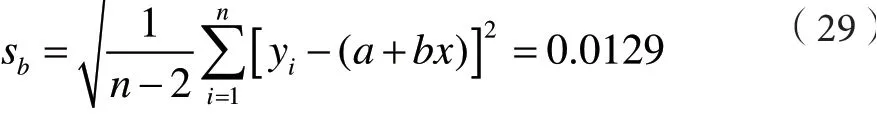
式(28)(29)中:b 为回归方程斜率;m 为试样中待测物含量的测定次数;n 为标准溶液的测定次数;x-为标准溶液浓度的平均值;x-0为待测物平均浓度;xi为由标准曲线方程得出的标准溶液的浓度测定值;yi为是相对于xi的测定值。标准曲线包含6 个浓度点,每个浓度点测定2 次,拟合曲线方程为y=1.23x-0.029 2,相关系数r=0.999 3 见表2,待测样品测定6 次的平均浓度值x-0=0.252 ng·mL-1,结果见表1。所以由标准曲线拟合引入的不确定度为:U(curve)=0.005 55,相对不确定度为:


表2 标准曲线拟合计算结果表
5.8 LC-MS/MS 仪器定量校准引入的不确定度
根据B 类评定、仪器检定证书,仪器定量重复性的标准偏差为3.7%,按均匀分布,仪器定量校准引入的不确定度为:

5.9 样品提取净化过程引入的不确定度
样品提取净化过程的不确定度用样品回收率的不确定度表示,选取回收率添加水平为0.25 µg·kg-1, 平行测定6 个加标回收样品,回收率(R)分别为98.8%、100.0%、97.6%、106.8%、95.6%以及105.2%, 平均回收率()为100.7%。回收率的标准偏差为:

根据A 类标准分布计算样品提取净化过程引入的不确定度为:

因此样品提取净化过程引入的不确定度:
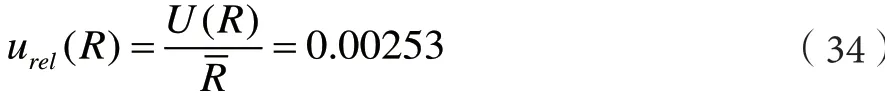
5.10 合成相对标准不确定度
根据以上分析及A 类评定方法,列出相对标准不确定度分量表(见表3)。

表3 各分量的相对标准不确定度分量表
合成的相对标准不确定度为:

鸡蛋中氯霉素残留量平均值为0.493μg·kg-1,因此测定过程中合成的相对标准不确定度为:
u(x)=urel(X)×=0.0412×0.493=0.0203 μg·kg-1(36)
扩展不确定度:选择置信度为95%,k=2,则扩展不确定度为U=0.020 3×2=0.040 6 μg·kg-1,鸡蛋中氯霉素残留量结果为(0.493±0.040 6)μg·kg-1。
6 结果与讨论
本实验使用液相色谱-串联质谱法对鸡蛋样品中氯霉素残留量进行了测定。结果显示,当鸡蛋样品中残留量为0.493 μg·kg-1时,其扩展不确定度为U=0.020 3×2=0.040 6 μg·kg-1。测量结果准确度较高,适用于鸡蛋中氯霉素的残留检测。通过表3 可知,实验过程中引入的不确定度对检测结果影响较大的主要分量有标准物质的配制、样品重复测定、标准曲线拟合、仪器定量校准;而标准物质称量、样品称重质量、最终定容体积、样品回收率等过程引入的不确定度分量对检测结果影响相对较小。因此在实验过程中应调整仪器使之达到最佳状态,制备样品时使样品充分均匀,确保实验结果更准确、稳定及可靠。
