基于高通量测序分析牛粪堆肥中细菌群落动态变化
2018-04-14许修宏成利军许本姝门梦琪张文浩邓利廷武晓桐盛思远
许修宏,成利军,许本姝,门梦琪,孙 瑜,张文浩,邓利廷,姜 欣,武晓桐,盛思远
(东北农业大学资源与环境学院,哈尔滨 150030)
近年我国畜禽养殖业快速发展,产生大量畜禽粪便,影响生态环境和人畜健康[1]。种植业方面,大量秸秆未有效利用,秸秆焚烧造成空气污染,资源浪费。好氧堆肥是对畜禽粪便和秸秆等有机固体废弃物作资源化、无害化有效处理方法[2],可将其转化为有机肥料,利于土壤改良和农业生态环境改善[3]。堆肥过程中细菌将有机大分子降解并释放热量,有害生物因子减少,实现堆肥无害化,形成腐殖质等稳定化合物[3]。堆肥是动态变化过程,细菌群落复杂,堆肥中细菌群落结构和动态研究受 到 关 注[4-5]。 常 采 用 T-RFLP[6]、 PCR-DGGE[7-9]、16S rRNA克隆文库[10]等方法分析堆肥过程中细菌群落结构,但存在难以检测低丰度细菌群落缺陷,16S rRNA基因高通量测序技术在测序深度上具有优势,可测是相对丰度0.01%~0.10%细菌群落及更多未命名微生物,增加数据库容量[11-12]。近年来,高通量测序技术逐渐应用于堆肥环境[13-14],de Gannes等利用高通量测序技术分析发现,在堆肥过程中升温期和腐熟期微生物群落不同,升温期存在的微生物在腐熟期未出现[15]。滑留帅等利用16S rRNA基因高通量测序技术分析牛粪自然发酵与添加益生菌剂发酵过程中细菌群落多样性变化,发现在牛粪自然发酵过程中细菌种群无明显变化规律,添加益生菌剂后,牛粪发酵过程中细菌种群不同于自然发酵[3]。
本研究通过高通量测序分析奶牛粪便和玉米秸秆堆肥过程中细菌群落结构变化及演替规律,对深入了解细菌在堆肥过程中作用及提高堆肥效果具科学价值。
1 材料与方法
1.1 堆肥原料和样品采集
堆肥材料为牛粪和玉米秸秆(见表1)。牛粪来自哈尔滨市幸福乡奶牛养殖场,玉米秸秆来自哈尔滨市香坊区东北农业大学实验实习基地。牛粪和玉米秸秆(粉碎为2 cm)按质量比5∶1均匀混合,初始含水率(Water content)为60%~65%,C/N为35∶1,置于规格为80×80×150 cm(长×宽×高)堆肥装置中,15 min通风1次,每次1 min,通风量为280 L·min-1,每日测定堆体温度和环境温度。堆肥过程持续36 d,取样时间为第1、2、3、4、7、11、14、19、25、36天,堆体上、中、下三层取样,每层平均取4个点,将所有样品均匀混合,每次取样后翻堆。堆肥样品分为两份:一份-4℃储存,测定理化指标;另一份-80℃储存,作细菌群落分析。

表1 堆肥原料理化参数Table 1 Physic-chemical parameters in compost raw materials
1.2 堆肥理化指标测定
采用精密数显电子温度计(LNI-TUT325)分别定时测定装置中堆体上、中、下3个位置温度及环境温度变化。含水率(Water content)用恒重法测定,烘干后样品用马弗炉在600℃下灼烧2 h测定有机物含量[16]。堆肥过程中有机物降解率(Organic matter degradation rate,OM)计算方程式:有机物降解率(%)=(堆肥初始有机物含量-第n天堆肥中有机物含量)/堆肥初始有机物含量×100%,其中有机物含量单位为kg·kg-1。将堆肥样品按1∶10(m/v)比例加入去离子水,震荡过滤后滤液用数字pH仪器测定pH。总有机碳(Total organic carbon,TC)和全氮(Total nitrogen,TN)分别采用重铬酸钾容量法和凯氏定氮法测定[17],两者比值即为碳氮比(C/N)。铵态氮(NH4+-N)和硝态氮(NO3--N)含量利用连续流动分析仪(SAN++SYSTEM,荷兰)测定。种子发芽指数(Germination index,GI)使用独行菜种子(Lepidium sativum L)测定[18]。将堆肥样品按1∶7.5(m/v)比例加入去离子水,取5 mL提取液滴入含有滤纸的培养皿中,将20粒饱满种子均匀分布在滤纸上,25℃黑暗条件下培养48 h。根据计算公式:GI(%)=处理平均发芽率×处理平均根长/(对照平均发芽率×对照平均根长)×100%,计算种子发芽指数。以上除温度外其他理化参数,每个样品均作3次重复测定。
1.3 MiSeq高通量测序
将原始样品(第1天)及升温期(第2天),高温期(第4、7、14天),降温期(第19天),腐熟期第36天样品送上海美吉生物医药科技有限公司Illumina Miseq PE300平台作高通量测序分析,记作C1、C2、C3、C4、C5、C6、C7。堆肥样品DNA提取使用 OMEGA 的 Soil DNA Isolation Kit(Omega Bio-Tek,Inc.,GA,USA),利用1%琼脂糖凝胶电泳检测抽提基因组DNA。对16S rRNA基因V3-V4高变区片段作PCR扩增,选择引物序列为338F-806R[19],338F(5'-ACTCCTACGGGAGGCAGCAG-3')和 806R(5'-GGACTACHVGGGTWTCTAAT-3')。扩增条件为:95℃预变性3 min,27个循环,包括95℃变性30 s,55℃退火30 s,72℃延伸45 s,循环结束后72℃延伸10 min。PCR产物用2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN)公司切胶回收PCR产物,Tris_HCl洗脱,用2%琼脂糖电泳检测,参照电泳初步定量结果,将PCR产物用QuantiFluor™-ST蓝色荧光定量系统(购自Promega公司)检测定量,按照每个样本测序量要求,按照相应比例混合,作Miseq测序。
1.4 生物信息处理
利用Mothur(V.1.36.1)对原始DNA序列作过滤处理,去除嵌合体,得优化序列;将相似性大于97%优化序列划分为一个操作分类单元(Operation⁃al Taxonomic Units,OTU),作稀释性曲线分析,计算Chao1丰度指数,覆盖度(Coverage)和Shannon多样性指数等。利用样本层级聚类分析和距离Heatmap分析各样品间OTU相似性。对比Silva 16S rRNA数据库(Release128 http://www.arb-silva.de),采用RDP分类器(Version 2.2 Release 11.1 http://rdp.cme.msu.edu/)贝叶斯算法对97%相似水平OTU代表序列作分类学分析,各分类水平上统计每个样品群落组成;利用相关性Spearman图分析堆肥样品理化因子与细菌群落结构关系。
1.5 数据统计分析
数据采用SPSS 21.0作统计分析。利用单因素方差分析法(ANOVA)作不同样品间差异显著性检验分析。
2 结果与分析
2.1 堆肥过程中理化参数变化
温度变化是堆肥过程重要参数[5]。堆肥过程中堆体和周围环境温度变化见图1,堆肥开始后堆体温度逐渐上升,第7天达最高温度65℃,后呈缓慢下降趋势,达环境温度。根据温度变化将堆肥过程划分为4个时期,即升温期(1~2 d)、高温期(3~14 d)、降温期(15~19 d)和腐熟期(25~36 d)。升温期,堆体含有大量易降解有机物,微生物快速分解有机质,产生CO2和水,释放热量,堆体温度迅速上升。高温期,大量嗜温微生物死亡或休眠,而嗜热微生物在降解有机质过程中具有重要作用。当堆体温度高于55℃时,进入高温期,持续5~7 d可杀死堆体中大量病原菌及有害物质[20]。本研究中堆体温度在第3天达55.6℃,标志堆体进入高温期,直到第14天,共持续12 d,符合粪便无害化卫生标准关于堆体温度要求。

图1 堆肥过程中的温度变化Fig.1 Change of temperature in cow manure composting
由表2可知,堆肥过程中,含水率整体呈下降趋势,腐熟期达48.80%;pH呈先升后降趋势,第4天达最大值8.85,变化范围为8.85~8.14,处于弱碱性状态,利于微生物保持良好活性[21]。碳氮比(C/N)是评价堆肥产品腐熟程度重要指标,C/N从初始值35.70下降到19.60,整体呈下降趋势。当C/N≤20.0时,认定堆肥达腐熟程度[22]。
有机物降解率是评价堆肥效果重要标志,反映堆肥腐熟降解过程[16]。由表2可知,有机物降解率整体呈上升趋势,堆肥前14 d有机物降解率为47.59%,堆肥结束后有机物降解率为55.17%,表明有机物降解主要发生在堆肥前期和高温期。Be⁃mal等发现堆肥化过程可使牛粪有机物降解率达60%,家禽粪便降解达52%,猪粪达72%[23],本研究中有机物降解率为55.17%,略低于60%,基本达到要求。种子发芽指数(GI)是指示堆肥腐熟程度的生物学指标,当GI≥80.0%时堆肥产品已经腐熟,植物毒性丧失[24]。
由表2可知,GI整体呈上升趋势,从堆肥前期28.30%升至86.20%,表明堆肥产品符合腐熟标准。Han等报道NH4+-N和NO3--N比值也可作为堆肥腐熟度评价指标,NH4+-N/NO3--N值小于1.0时可认定堆肥已腐熟[25]。
由表2可知,NH4+-N/NO3--N值呈下降趋势,第25天达0.90,第36天达0.51,可认定堆肥已腐熟。根据以上指标综合判断,本研究中利用堆肥装置处理牛粪和玉米秸秆混合物,堆肥36 d可达腐熟要求。
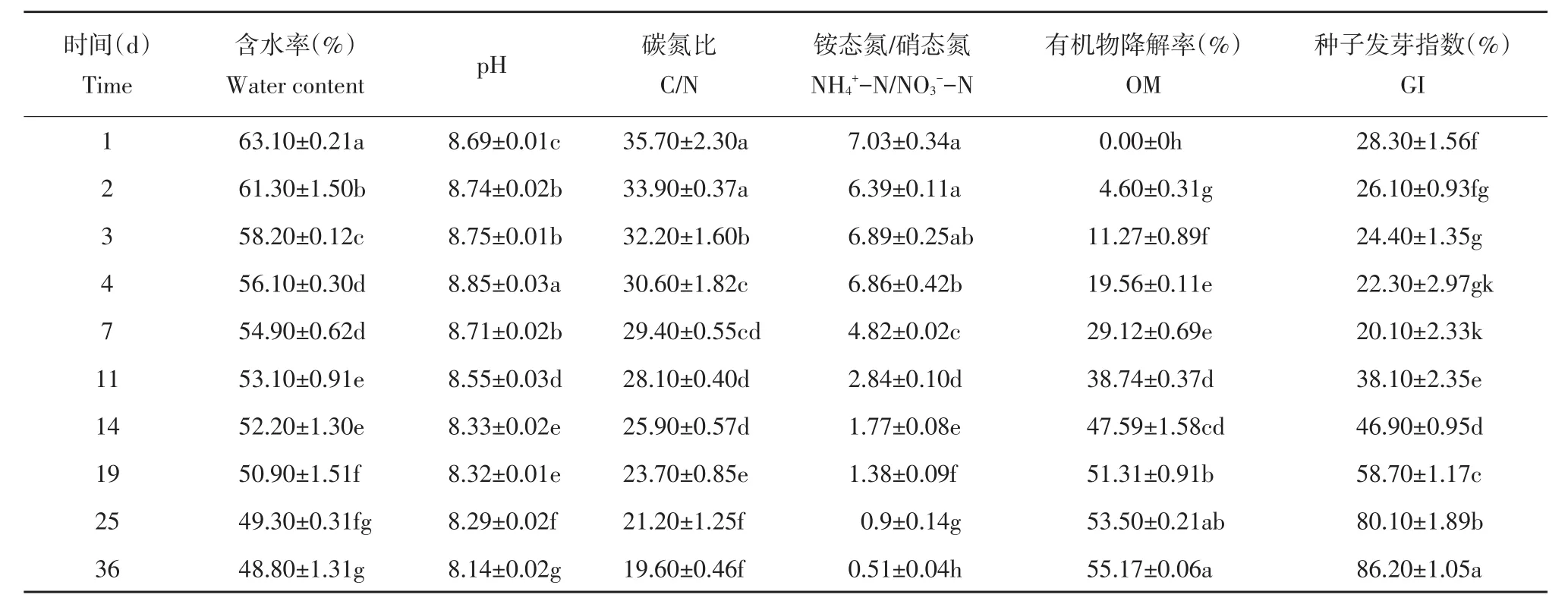
表2 堆肥过程中理化性质变化Table 2 Changes in physic-chemical properties in cow manure composting
2.2 堆肥中细菌群落丰度、多样性和结构差异
所有堆肥样品通过高通量测序共获得274 759条高质量序列,依次为34 881、40 862、38 112、43 382、42 763、39 202和35 557条。为更好比较分析,所有样品序列标准化为18 182条。表3分别是堆肥样品优化序列、OTU数量及多样性指数。对标准化序列基于97%相似度水平划分,共获得1 001个OTUs。由表3可知,堆肥过程中7个样品被检测OTU数量先逐渐增加,C4达最高值为542,之后依次减少,C7 OTU数量最少为179个,说明本次堆肥中高温期细菌群落丰度最高。本研究通过分析各样品文库覆盖率(Coverage)和稀释曲线说明测序深度。表3中各样品文库覆盖率均超过99.3%,说明堆肥样品中基因序列被检概率较高;图2是各样品OTUs稀释曲线,随机抽样序列,以抽到序列数与其所代表OTU数目构建稀释曲线,曲线趋向平坦时,说明测序数据量合理,更多数据量会产生少量新OTU,反之则表明继续测序还可能产生较多新OTU,由图2可知各样品稀释曲线均趋于平坦,表明本次测序深度满足分析要求。综合各样品文库覆盖率和稀释曲线认为本次测序结果可代表堆肥中细菌群落真实情况。
Alpha多样性被称为生境内多样性,研究局域均匀生境下物种数目[26],堆肥过程中细菌Alpha多样性统计结果见表3。
Chao1指数用于估算样品中所含OTU数目指数,反映细菌菌群丰富度,Chao1指数越大说明细菌菌群丰富度越高。Simpson指数是估算样品中微生物多样性指数之一,Simpson指数值越大,说明微生物群落多样性越低。Shannon指数也是估算样品中微生物多样性指数之一,Shannon值越大,说明微生物群落多样性越高。
由表3可知,在堆肥过程中,Chao1指数呈先增后减趋势,最大Chao1指数出现在高温期(C3、C4和C5),其次是升温期C2和原始样品C1,降温期C6和腐熟期C7最少,说明高温期堆肥中细菌菌群丰富度最高。Shannon和Simpson指数变化基本一致,细菌群落多样性由高到低为C1>C4>C2>C5>C3>C6>C7。

图2 各样品OTUs稀释曲线Fig.2 Rarefaction curvesfor OTUsof each sample

表3 堆肥样品中细菌丰度与多样性Table 3 Bacterial richnessand diversity in composting samples
根据7个堆肥样品中OTU组成作样本层级聚类分析(见图3A)和样品之间距离Heatmap图分析(见图3B),在图3B中样本间距离用一定颜色梯度表示(图中右侧为颜色梯度代表数值),颜色越深,样本之间距离越大,表示细菌群落结构差异性越大。
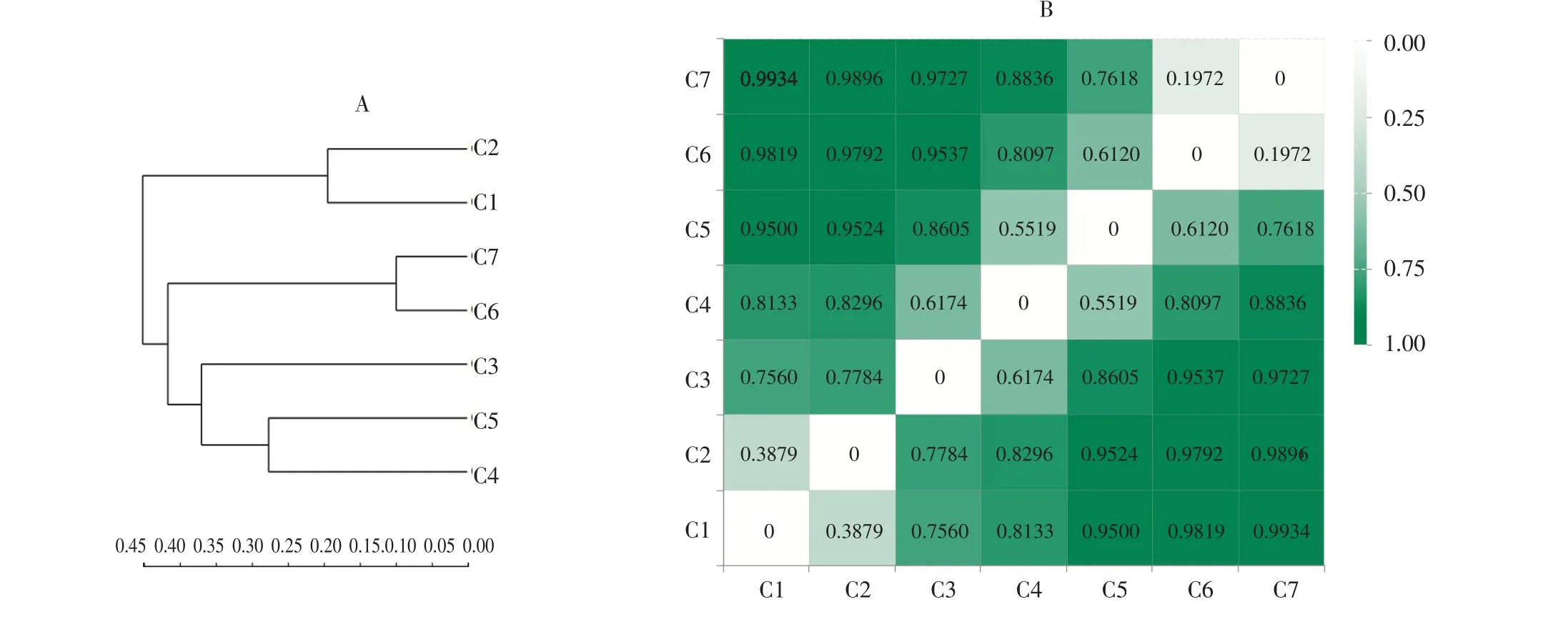
图3 堆肥样本层级聚类树(A)和距离Heatmap(B)Fig.3 Hierarchical clustering tree and distances heatmap on OTU level of composting samples
由图3可知,降温期C6和腐熟期C7之间距离最近,为0.1972,即两者OTUs组成相近;原始样品C1与升温期C2之间距离次之,为0.3879,即两者OTUs组成相似;高温期C4与高温期末期C5 OTU组成相近,为0.5519,但差异性大于C1与C2;由图3-B可知,高温初期C3与C1、C2、C5、C6和C7之间距离较大,与C4最近,为0.6174,即两者之间细菌群落结构差异性大于C4和C5;堆肥前期、高温期及腐熟期样本之间细菌群落结构差异性最大,高温期和腐熟期差异性次之。
2.3 堆肥中细菌门水平分类和属水平分类
7个堆肥样品中1 001条OTUs分属于23个门,51个纲,112个目,232个科,418个属,607个种。图4A为门类水平细菌分类(将相对丰度<1.0%部分合并为others在图中显示),主要包括酸杆菌门(Acidobacteria),变形菌门(Proteobacteria),厚壁菌门(Firmicutes),拟杆菌门(Bacteroidetes),绿弯菌门(Chloroflexi),放线菌门(Actinobacteria)。堆肥过程中,7个堆肥样品门分类细菌相对丰度存在一定变化趋势。
酸杆菌门(Acidobacteria)相对丰度呈逐渐上升趋势,首次出现在升温期(C2),其相对丰度为0.3%,腐熟期(C7)达最高93.9%,成为腐熟期优势类群,变形菌门相对丰度呈先降后升再降趋势,主要存在于堆肥前期和高温期,在堆肥初始阶段(C1,55.3%)、升温期(C2,45.5%)、高温期初期(C3,74.3%)和高温期(C4,33.6%)始终是优势类群,高温期末期(C5)相对丰度也高达22.3%,是此时期第二高的门。降温期和腐熟期,变形菌门相对丰度较少,分别为6.7%和1.4%;厚壁菌门相对丰度整体呈先升后降趋势,主要存在于升温期(C2,24.8%)和高温期(C3,15.6%;C4,30.7%;C5,12.8%),高温期(C4)相对丰度最高,也是该样品中相对丰度第二高的门;拟杆菌门相对丰度呈下降趋势,主要存在堆肥初始阶段(C1,28.5%)和升温期(C2,19.4%),堆肥后期存在波动,基本处于1.2%~6.4%,主要因拟杆菌对碳水化合物具有较好降解能力[27];绿弯菌门相对丰度呈先升后降趋势,主要出现在高温期和降温期,在高温期末期相对丰度达到最高值为19.2%,Fracchia等在堆肥降温期和腐熟期检测到绿弯菌门细菌[28];放线菌门相对丰度变化趋势不明显,变化范围为0.5%~8.5%,整个堆肥过程始终存在,高温期(C4,8.5%)相对丰度最高,放线菌门通过分泌各种抗生素抑制甚至杀死病原微生物,有利于降低堆肥产品生物毒性[4]。

图4 堆肥样品中在门(A)和属(B)分类水平上细菌种类和相对丰度Fig.4 Relativeabundance of bacterial during composting processat thelevel of phylum(A)and genus(B)
图4 B为属分类水平上细菌分类(将相对丰度<5.0%部分合并为others在图中显示),剩余11个属水平分类中有一个属为分类学数据库分类学谱系中间等级,无科学名称,以norank作为标记。优势类群主要包括:类似芽球菌属(Blastocatella)、肠杆菌属(Enterobacter)、环脂酸芽孢杆菌属(Tumebacil⁃lus)、玫瑰弯菌属(Roseiflexus)、未命名细菌(norank_c_OPB54)、黄杆菌属(Flavobacterium)、戴沃斯菌属(Devosia)、假黄色单胞菌属(Pseudox⁃anthomonas)、假单胞菌属(Pseudomonas)、土地杆菌属(Pedobacter)、金黄杆菌属(Chryseobacterium)。C1中主要含有肠杆菌属(10.4%)、黄杆菌属(7.6%)、假单胞菌属(6.3%)、戴沃斯菌属(5.9%)、土地杆菌属(5.2%)、金黄杆菌属(5.1%)和环脂酸芽孢杆菌属(0.4%),其中黄杆菌属是拟杆菌门,促进多糖类分解,存在于堆肥前期[27];假单胞菌属是变形菌门,主要在堆肥前期发挥作用可降解蛋白质和淀粉等有机物,不耐高温,当温度高于40℃其生物活性降低[29],从图4B可知,进入高温期后假单胞菌属相对丰度逐渐降低。张晶等利用牛粪作为原料堆肥时,在堆肥前期检测到大量假单胞菌属,但经高温期后其相对丰度逐渐降低[16]。在堆肥升温期(C2)出现大量环脂酸芽孢杆菌属(21.3%),为优势菌群,随堆体温度逐渐升高其相对丰度逐渐,高温期初期C3环脂酸芽孢杆菌属相对丰度仅0.31%,高温期末期C5相对丰度仅0.06%,降温期C6和腐熟期C7未发现,主要存在于堆肥前期;肠杆菌属(7.6%)、黄杆菌属(5.8%)、戴沃斯菌属(5.8%)、假单胞菌属(2.4%)、土地杆菌属(3.4%)和金黄杆菌属(1.4%)相对丰度下降,除肠杆菌属,其他5个菌属随着堆体温度升高,在高温期大幅度降低。在堆肥高温期初期C3肠杆菌属相对丰度最高为47.9%,类似芽球菌属、未命名细菌(norank_c_OPB54)和假黄色单胞菌属相对丰度分别为1.0%、2.3%和9.0%,其中假黄色单胞菌属是此阶段相对丰度第二高的属。在堆肥高温期C4未命名细菌(norank_c_OPB54)成为相对丰度最高的属,为18.6%,肠杆菌属相对丰度为14.2%,与C3比较大幅度降低,类似芽球菌属(9.5%)增多,成为高温期相对丰度第三高属,玫瑰弯菌属首次出现该堆肥中,相对丰度为3.4%,假黄色单胞菌属(2.5%)和假单胞菌属(0.8%)相对丰度均降低。在堆肥高温期末期C5,类似芽球菌属(26.1%)继续升高,成为此阶段相对丰度最高属,玫瑰弯菌属大量增多,相对丰度达14.4%,未命名细菌(norank_c_OPB54)相对丰度则减少为2.3%。在降温期C6和腐熟期C7优势菌群分布结构相似度较高,且比较单一,主要类似芽球菌属,相对丰度分别为72.6%和93.4%,在降温期玫瑰弯菌属相对丰度为3.97%,假单胞菌属和未命名细菌(norank_c_OPB54)相对丰度均特较少,分别为0.11%和0.23%。
2.4 堆肥中细菌群落结构与环境因子关系
选取前15个优势属(至少在一个样品中相对丰度>1.0%)统计分析与环境因子间相关性,作属水平堆肥中主要细菌群落结构与环境因子Spearman相关性,如图5所示。结果表明除肠杆菌属、未命名细菌(norank_c_OPB54)、假黄色单胞菌属外,剩余11个属均与含水率、有机物降解率、C/N和NH4+-N/NO3--N之间呈显著相关(P<0.05),甚至极显著相关(P<0.001);其中类似芽球菌属与含水率、C/N和NH4+-N/NO3--N之间呈极显著负相关(P<0.001);肠杆菌属与pH呈极显著正相关(P<0.001);藤黄单胞菌属(Luteimonas)、短波单胞菌属(Brevundimonas)、叶杆菌科未分类细菌(unclassi⁃fied_f_Phyllobacteriaceae)、环脂酸芽孢杆菌属和戴沃斯菌属与pH呈显著正相关(P<0.05)。环境因子温度(Temp)仅与未命名细菌(norank_c_OPB54)呈极显著正相关(P<0.001),与图4B相符,该菌是嗜热细菌,耐高温。类似芽球菌属与pH P值为-0.052,表明两者相关性不显著。

图5 Spearman相关性热图Fig.5 Spearman correlation heatmap
3 讨论与结论
利用高通量测序技术分析牛粪和玉米秸秆混合堆肥过程中细菌群落动态变化,统计分析表明,堆肥高温期细菌群落丰度最大,不同时期堆肥样品细菌多样性指数存在显著差异,由高到低依次是堆肥前期>高温期>降温期和腐熟期。Ren等利用牛粪作为原料堆肥时,堆肥过程中细菌群落多样性由高到低为升温期>堆肥初期>高温期>降温期和腐熟期,与本研究相符[5]。堆肥前期细菌群落多样性高,主要是堆肥前期细菌可利用底物质丰富和温度适合生存,物种较多,进入高温期后,随着温度升高,大量嗜热细菌繁殖,不耐高温细菌减少,多样性降低,堆肥降温期和腐熟期细菌可利用底物减少,大部分底物不易降解,细菌群落多样性降低[5]。将各堆肥时期样品OTU组成作样本层级聚类分析和样品之间Heatmap图分析表明,堆肥前期细菌群落结构变化显著,高温期变化最大,降温期和腐熟期细菌群落结构差异性最小。张晶等报道堆肥前期微生物群落结构变化显著,堆肥后期保持稳定,与本研究结果一致[16]。陆彦宇利用牛粪和中药渣堆肥发现,细菌经过堆肥高温期阶段前后种群发生较大变化[30]。门分类水平细菌群落结构分析表明,变形菌门和厚壁菌门在堆肥前期和高温期相对丰度较高,堆肥后期逐渐降低,酸酐菌门在堆肥后期相对丰度逐渐升高。变形菌门是细菌中最大一门,在堆肥过程中具有重要作用[3],厚壁菌门可在高温环境下存活,与各种代谢活动有关,主要出现在堆肥高温期[4-5],陆彦宇等研究发现,在堆肥过程中变形菌门和厚壁菌门微生物逐渐减少,与本研究相符[30];酸杆菌门细菌一般是一类嗜酸、寡营养和难培养菌类,但实际上有些酸杆菌可在中性甚至碱性环境中被检测[31],其主要功能是降解植物残体[32],本研究中堆肥后期整体生态环境呈弱碱性,底物质成分主要为难降解玉米秸秆,因此推测本研究中酸杆菌门是一类可在弱碱性环境中降解植物残体的菌群。
属分类水平细菌群落结构分析表明,堆肥初始阶段各属相对丰度相近,环脂酸芽孢杆菌属在堆肥升温期占优势,肠杆菌属、未命名细菌(norank_c_OPB54)和玫瑰弯菌属为堆肥高温期优势菌群,类似芽球菌属在降温期和腐熟期成为优势菌属。环脂酸芽孢杆菌属主要分解有机质,是一类利用碳源细菌[33],堆肥前期存在大量易降解有机质,因此环脂酸芽孢杆菌属较多。肠杆菌属是变形菌门中γ-变形菌纲肠杆菌科,主要存在堆肥前期和高温期,可提高堆体发酵温度,加快有机质分解速度,利于降低堆体含水率[34]。未命名细菌(norank_c_OPB54)属于厚壁菌门,最先从厌氧环境中分离,对木质纤维素分解作用较强[35-36],从升温期到高温期可能因微生物反应过于剧烈,易降解有机物被快速分解,氧气消耗较多形成局部缺氧导致该细菌大量产生。玫瑰弯菌属为一类嗜热菌属,主要存在牛粪堆肥高温期[16]。类似芽球菌属属于酸杆菌门,酸杆菌是土壤中一类重要细菌类群,王光华等基于16S rRNA基因序列分析发现,酸杆菌一般占细菌总量20%,甚至高达50%以上,表明酸杆菌在土壤生态过程中起重要作用[32],但在堆肥环境中酸杆菌报道较少。堆肥后期酸杆菌对堆肥发酵影响及其在堆肥生态过程中作用,需进一步研究。
Spearman相关性分析表明,堆肥过程中细菌群落结构变化与环境因子显著相关,其中未命名细菌(norank_c_OPB54)与温度呈极显著正相关(P<0.001),类似芽球菌属与含水率、有机物降解率、C/N和NH4+-N/NO3--N之间呈显著相关,与pH相关性不显著,类似芽球菌属属于酸杆菌门。研究结果表明,酸杆菌相对丰度与pH呈显著负相关[37-38]。
[参 考 文 献]
[1] 尹昌斌,周颖,刘利花.我国循环农业发展理论与实践[J].中国生态农业学报,2013,21(1):47-53.
[2] 刘佳,许修宏,李洪涛.利用PCR-DGGE技术对自然堆肥微生物群落多样性的分析[J].东北农业大学学报,2009,40(9):35-38.
[3] 滑留帅,王璟,徐照学,等.16SrRNA基因高通量测序分析牛粪发酵细菌多样性[J].农业工程学报,2016,32(增2):311-315.
[4] Tian W,Sun Q,Xu D,et al.Succession of bacterial communities during composting process as detected by 16SrRNA clone librar⁃ies analysis[J].International Biodeterioration and Biodegradation,2013,78(2):58-66.
[5] Ren G,Xu X,Qu J,et al.Evaluation of microbial population dy⁃namics in the co-composting of cow manure and rice straw using high throughput sequencing analysis[J].World Journal of Microbi⁃ology and Biotechnology,2016,32(6):1-11.
[6] Tiquia SM.Using terminal restriction fragment length polymor⁃phism(T-RFLP)analysis to assess microbial community struc⁃ture in compost systems[J].Methods in Molecular Biology,2010,599:89-102.
[7] Wang SL,Liu Y Q,Liu Y,et al.PCR-DGGE analysis of the bac⁃terial community in composting of agriculture and forestry wastes with different microbial agents[J].Advanced Materials Research,2014,1010(4):966-972.
[8] Asano R,Otawa K,Ozutsumi Y,et al.Development and analysis of microbial characteristics of an acidulocomposting system for the treatment of garbage and cattle manure[J].Journal of Biosci⁃enceand Bioengineering,2010,110(4):419-425.
[9] Knerr A,Tripepi R R.Changes in bacterial communities in dairy manure during nine months of composting as determined by dena⁃turing gradient gel electrophoresis[J].International Environmental Agreements Politics Lawand Economics,2012,12(4):327-342.
[10] Lee Y H,Kim S K,Yong H K,et al.Archaeal diversity during composting of pig manure and mushroom cultural waste based on 16SrRNA sequence[J].Journal of the Korean Society for Applied Biological Chemistry,2010,53(2):230-236.
[11] Chu Z R,Wang K,Li X K,et al.Microbial characterization of aggregates within a one-stage nitritation-anammox system using high-throughput amplicon sequencing[J].Chemical Engineering Journal,2015,262:41-48.
[12] Petrosino JF,Highlander S,Luna RA,et al.Metagenomic pyrose⁃quencing and microbial identification[J].Clinical Chemistry,2009,55(5):856.
[13] Cui E,Wu Y,Zuo Y,et al.Effect of different biochars on antibiotic resistance genes and bacterial community during chicken manure composting[J].Bioresource Technology,2016,203(4):11-17.
[14] Tkachuk V L,D.O.Krause K,Knox N C,et al.Targeted 16S rRNA high-throughput sequencing to characterize microbial com⁃munities during composting of livestock mortalities[J].Journal of Applied Microbiology,2014,116(5):1181-1194.
[15] De Gannes V,Eudoxie G,Hickey W J.Prokaryotic successions and diversity in composts as revealed by 454-pyrosequencing.[J].Bioresource Technology,2013,133(2):573.
[16] 张晶,孙照勇,钟小忠,等.奶牛粪条垛式模拟堆肥腐熟度及微生物群落结构变化[J].应用与环境生物学报,2016(3):423-429.
[17] 鲍士旦.土壤农化分析[M].北京:中国农业出版社,2008.
[18] Zucconi F,Pera A,Forte M,et al.Evaluating toxicity of immature compost[J].Biocycle,1981,22(2):54-57.
[19] Dennis K L,Wang Y,Blatner N R,et al.Adenomatous polyps are driven by microbe-instigated focal inflammation and are con⁃trolled by IL-10 producing T-cells[J].Cancer Research,2013,73(19):5905-5913.
[20]中国预防医学科学院环境卫生与卫生工程研究所.GB7959—87粪便无害化卫生标准[S].北京:中国标准出版社,1988.
[21] Mas M A,Blasi A B.Evaluation of composting as a strategy for managing organic wastes from a municipal market in Nicaragua[J].Bioresource Technology,2008,99(11):5120-5124.
[22] 栾冬梅,关静姝,徐瑨,等.碳氮比对牛粪好氧堆肥过程的影响[J].东北农业大学学报,2008,39(8):77-81.
[23] Bernal M P,Alburquerque J A,Moral R.Composting of animal manures and chemical criteria for compost maturity assessment.A review[J].Bioresource Technology,2009,100(22):5444-5453.
[24]Wang X,Selvam A,Chan M,et al.Nitrogen conservation and acid⁃ity control during food wastes composting through struvite forma⁃tion[J].Bioresource Technology,2013,147(8):17-22.
[25]Han JK,Kim K Y,Kim H T,et al.Evaluation of maturity parame⁃ters and heavy metal contents in composts made from animal ma⁃nure[J].Waste Management,2008,28(5):813-20.
[26] 常安然,李佳,张耸,等.基于宏基因组学16SrDNA测序对烟草根际土壤细菌群落组成分析[J].中国农业科技导报,2017,19(2):43-50.
[27] Martin M,Barbeyron T,Martin R,et al.The Cultivable surface microbiota of the brown alga Ascophyllum nodosumis enriched in macroalgal-polysaccharide-degrading bacteria[J].Frontiers in Microbiology,2015,6:1-13.
[28] Fracchia L,Dohrmann A B,Martinotti M G,et al.Bacterial diver⁃sity in a finished compost and vermicompost:Differences revealed by cultivation-independent analyses of PCR-amplified 16S rRNA genes[J].Applied Microbiology and Biotechnology,2006,71(6):942-952.
[29] 尚晓瑛,程旭艳,霍培书,等.1株堆肥耐低温纤维素降解菌的筛选、鉴定及生长特性的初步研究[J].华中农业大学学报:自然科学版,2012,31(5):558-562.
[30]陆彦宇.牛粪和中药渣堆肥化过程中水溶有机质特性及细菌结构的研究[D].南宁:广西大学,2017.
[31] Xiong J,Liu Y,Lin X,et al.Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau[J].Environmental Microbiology,2012,14(9):2457-2466.
[32] 王光华,刘俊杰,于镇华,等.土壤酸杆菌门细菌生态学研究进展[J].生物技术通报,2016,32(2):14-20.
[33] 杜思瑶,于淼,刘芳华,等.设施种植模式对土壤细菌多样性及群落结构的影响[J].中国生态农业学报,2017,25(11):1615-1625.
[34] 徐庆贤,官雪芳,林碧芬,等.几株猪粪堆肥发酵菌对堆肥发酵的促进作用[J].生态与农村环境学报,2013,29(2):253-259.
[35] Wong M T,Wang W,Lacourt M,et al.Substrate-driven conver⁃gence of the microbial community in lignocellulose-amended enrichmentsof gut microflorafrom thecanadian beaver(Castor ca⁃nadensis)and North American Moose(Alces americanus)[J].Fron⁃tiers in Microbiology,2016,7:961-970.
[36] Hao L P,Lv F,Mazeas L,et al.Stable isotope probing of acetate fed anaerobic batch incubations shows a partial resistance of ace⁃toclastic methanogenesis catalyzed by Methanosarcina to sudden increaseof ammonialevel[J].Water Research,2015,69:90-99.
[37] Griffiths R I,Thomson B C,James P,et al.The bacterial biogeog⁃raphy of British soils[J].Environmental Microbiology,2011,13(6):1642.
[38] Jones R T,Robeson M S,Lauber C L,et al.A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clonelibrary analyses[J].Isme Journal,2009,3(4):442-453.
