Keggin型多酸负载的单原子催化剂(M1/POM, M = Ni, Pd, Pt, Cu,Ag, Au, POM = [PW12O40]3-)活化氮气分子的密度泛函理论计算研究
2018-04-10尹玥琪蒋梦绪刘春光
尹玥琪,蒋梦绪,刘春光
1 引言
氮元素是自然界中十分重要的元素之一,它是植物生长,人类生产、生活必不可少的基本元素。由于氮元素在自然界中主要以氮气分子的形式存在,而氮气的化学性质又十分稳定,因此不能被动植物直接吸收和利用。自然界中主要依靠闪电,或者植物中的固氮菌来实现氮元素的转化1,2。随着工、农业生产对氮元素需求量的不断增加,仅仅依靠生物固氮已经远远不能满足现代社会发展的需要。因此,常温、常压下人工固氮的方法被各界学者广泛关注。采用化学方法,制备出类似植物“固氮菌”的物质,使空气中的氮在常温常压下就能转变为铵态氮或氨态氮供人们利用,成为学界关注的前沿热点问题之一3-8,特别是双氮配位化学,由于在人工固氮方面的潜在应用而特别受到关注9,10。在过去的半个多世纪里,许多学者认为具有潜在固氮作用的过渡金属-氮气配合物具有活化氮气分子的能力11-13。目前已经报道了许多具有新颖化学结构的过渡金属-氮气配合物14,15。其中Chatt循环是这一领域最具代表性的范例之一(图1)。该循环基于一个单核钼-氮气膦配合物,通过连续的多步质子耦合电子转移,最终将一个配位的氮气分子转化为两个氨气分子,而单核钼膦配合物在循环过程中起到了催化剂的作用。
充分应用负载型金属催化剂每一个原子的最有效的方法是缩小金属体相材料的尺寸到单个的独立原子,含有孤立的单原子作为活性中心的负载型金属催化剂被定义为单原子催化剂(SACs)。由于SACs是由孤立的单金属原子分散在负载材料表面组成,因此,从理论上说,分散度应该是100%。这样的催化剂与传统的负载型金属纳米粒子催化剂是截然不同的。在过去的几年里,有很多例子毋庸置疑的证明了负载型SACs优异的催化行为。SACs不但表现出催化活性而且具有最高的催化活性,并且在催化反应过程中十分的稳定。这主要是由于金属单原子与载体表面锚定位点之间具有较强的键连作用16-19。例如:对于CO的氧化和优先氧化,Pt1/FeOx单原子催化剂的活性是亚纳米尺寸催化剂活性的2-3倍,并且,在较长的检测周期内都具有很高的稳定性16。对于3-硝基苯乙烯的加氢反应,Pt1/FeOx单原子催化剂的周转频率达到了1500 s-1,是目前报道的最好结果的20倍左右20。另外,对于氨基苯乙烯的加氢反应,该催化剂的选择性达到了99%,这是目前Pt族催化剂获得的最大值。将Pd单原子分散到乙烯醇盐的表面,乙烯醇盐被稳定在超薄的TiO2纳米片上。通过此方法合成的Pd1/TiO2单原子催化剂在C=C键加氢反应中表现出了很高的催化活性,超过市售Pd催化剂表面Pd原子的九倍。乙醛加氢催化反应增加了55倍21。孤立的单金属原子分散在恰当的载体表面表现出的优异的催化活性打开了通往多相催化前沿的大门。

图1 单核钼膦配合物的固氮Chatt循环Fig.1 The chatt cycle for nitrogen fixation on a mononuclear molybdenum-phosphine complex.
多酸(POMs)全称多金属氧酸盐,是一类由氧原子桥接金属原子形成的金属-氧簇化合物,由于它具有超强的酸性和优良的氧化还原性,成为一种新型的兼具酸和氧化还原催化活性的双功能催化剂。POMs类化合物催化活性高,选择性好,对设备腐蚀性小,同时又不产生污染,是一种环境友好和很有发展前途的绿色催化剂,目前已经被广泛应用于酯类水解、烯烃环氧化、烷烃羟基化,光催化分解水等一些意义重大的反应中22-30。不同于金属氧化物,POMs的体相材料由孤立的阴离子基团与抗衡离子构成,而表现为不连续的结构。更形象的说,POMs类化合物更像是由独立的金属氧化物“碎片”组成。早在1970年代,这类化合物就被选择作为催化剂载体材料31。最近,以Keggin型POM为载体的SACs(Pt1/POM,POM =[PW12O40]3-),已经被成功合成32,33,并且Pt1/POM催化剂在芳烃加氢反应中表现出了优异的催化活性和选择性32。
本文采用密度泛函理论(DFT)计算方法探究了一系列以多酸为载体的 SACs (M1/POM (M =Ni, Pd, Pt, Cu, Ag, Au, POM = [PW12O40]3-)潜在的活化氮气分子的能力。本文的主要目的在于探讨研究体系的电子结构和金属-氮气成键相互作用与活化氮气之间的内在联系,为后续的氮转移反应做好前期理论工作。
2 计算细节
所有的分子几何构型都采用 meta-广义梯度近似(meta-GGA)的交换相关泛函 M06L进行优化。M06L泛函采用一种平衡的方法将自旋能量密度分配到交换和相关泛函中。前期的理论研究工作已经证明 M06L泛函在计算由主族和过渡金属元素组成化合物的热化学、动力学、非共价键相互作用和频率等都表现出了较好的行为34。同时,理论计算表明 M06L泛函在预测多酸化合物的分子几何结构、振动频率和热化学等性质都表现出了较高的准确性30。主族元素采用双ζ层劈裂价基加极化函数6-31G(d)来构筑分子的哈密顿量,考虑到相对论效应,金属元素采用LANL2DZ基组35,36。为了确定计算的分子结构是势能面上的极小值点和获得热力学性质,在相同的理论水平上进行了频率计算。溶剂化效应通过采用SCRF(自洽场反应)方法的 IEFPCM(积分方程形式的连续极化模型)进行计算37。基于优化的分子几何结构,采用自然键轨道理论分析(NBO)38程序检验所研究化合物的电子结构。所有的计算都是通过Gaussian 09程序包完成39。
N2的吸附能计算公式为:

Εcomplex是金属-氮气 POM 配合物的能量,ΕM-POM是过渡金属-氮气配合物的能量,ΕN2是自由氮气分子的能量。
M1/POM 的过渡金属与多酸载体之间的吸附能计算公式为:
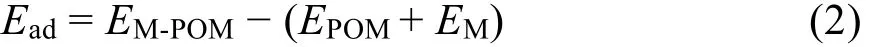
ΕM-POM是POM的能量,ΕPOM是POM的能量,ΕM是游离金属原子的能量。
3 结果与讨论
过渡金属原子往往会依据不同的配位环境而表现出不同的自旋态。根据实验和理论计算表明,在以多酸为载体的M1/POM体系中孤立的金属原子往往锚定在多酸表面桥氧原子组成的四重洞位上(见图2)40。我们采用DFT-M06L计算了本文中每个体系所有可能自旋态的总能量(总能量包括电子能量和零点校正能量),计算结果列于表S1(见Supporting Information)中。对于处于同一族的Ni、Pd、Pt三种金属原子,它们都具有d8电子组态,在原子的5个d轨道上有两种排布形式,因此,每个体系都可能具有单重和三重两个自旋态。对于处于同一族的 Cu、Ag、Au三种金属原子具有d9电子组态,在原子的5个d轨道上只有一种排布形式,所以,每个体系只能具有一个二重态。我们的计算结果表明前过渡金属 Ni1/POM体系具有一个高自旋的三重基态,后过渡金属Pd1/POM和Pt1/POM体系具有低自旋的单重基态。众所周知,前过渡金属原子往往以高自旋态最稳定30。相反,后过渡金属原子主要以低自旋态能量最低。我们的DFT-M06L计算结果和这一结论是完全一致的。
锚定于多酸表面四重洞位的独立金属原子与配位的氧原子构成了一个四棱锥的几何结构,金属原子位于四棱锥的顶点上(见图 2)。通过DFT-M06L计算得到的M1/POM体系中金属原子与四个配位的氧原子之间的平均距离avM―O列于图2中。计算结果表明avM―O值按照Ni1/POM <Cu1/POM < Ag1/POM < Pd1/POM < Au1/POM <Pt1/POM 的顺序递增。Ni1/POM 体系给出了小的avM-O值。采用M06L计算获得的单金属原子在多酸表面的吸附能(单位均为 kJ·mol-1)按照Ni1/POM (-448.02)< Pt1/POM (-133.34)<Cu1/POM (-116.06)< Pd1/POM (-111.29)<Ag1/POM (-111.04) < Au1/POM (-108.78)顺序升高,Ni1/POM 体系给出了最负的吸附能。大量的实验研究表明负载型金属催化剂中金属-载体之间相互作用不宜过强。金属-载体之间相互作用过强势必会消弱中心金属原子与反应物分子之间的相互作用,从而降低其催化活性。对于本文研究的体系,金属-载体之间相互作用最强的Ni1/POM多酸体系的催化活性将较弱。
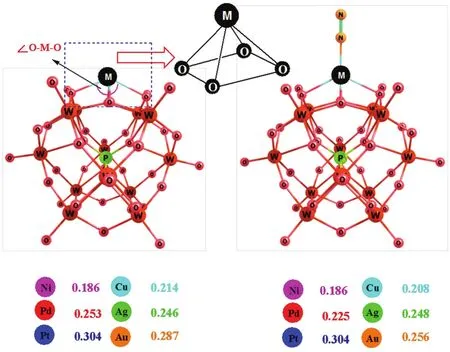
图2 M1/POM体系吸附氮气前后的分子几何结构和金属与四个配体氧的平均键长(avM-O, nm)Fig.2 Molecular structure of M1/POM and nitrogen-metal complex and average bond length (avM-O, nm).
值得注意的是Pt1/POM体系也具有较负的吸附能。这主要由于是Pt1/POM的结构中锚定于多酸表面四重洞位的Pt原子与配位的氧原子构成的四棱锥结构并非是正四棱锥,Pt原子向其中的一个氧原子偏移,这一现象是导致Pt1/POM有较大avM-O值的同时还具有较负吸附能的原因,这一特殊结构直接导致它具有较强的活化氮气的能力。
为了验证对这些分子几何结构上的猜测,对氮气分子在SACs上的吸附能进行计算(金属原子采用LANL2DZ基组),吸附前后的结构模型在图2中比较列出。计算出的数据也都列在表1中,可发现吸附能按照 Pt1/POM < Pd1/POM< Cu1/POM <Ag1/POM < Au1/POM < Ni1/POM 的顺序递增。尤其是Pt1/POM吸附能最负,达到-239.16 kJ·mol-1,这个结果与之前分子几何结构的预测完全一致。
为了分析POM载体在SAC体系中的作用,分别计算了氮气分子在Pt1/POM、孤立的Pt原子和两种由八个 Pt原子构成的团簇(Pt8)上的吸附能。相关结果列于表S2中。计算结果表明,以多酸为载体 Pt1/POM体系 N2吸附能与独立金属 Pt原子相当。但是,比上述两种团簇更负。这说明孤立的Pt原子相比于Pt原子团簇有着更加优异的氮气吸附特性,POM 载体没有影响到独立的 Pt原子对氮气分子的吸附性能。
吸附氮气前后分子几何结构和相关参数比较于图2中,计算结果表明,由于无机POM笼的刚性结构,在吸附氮气前后,POM载体部分几何结构参数基本没有发生明显改变。氮气吸附配合物的关键分子几何结构参数列于表 1中。表中数据表明,M―N 键长按照 Pt < Cu < Pd < Au < Ag < Ni的顺序递增,优化后 N≡N键距按照 Ni < Ag < Cu <Au < Pd < Pt的顺序递增。为了进一步探究负载金属单原子与氮气之间的相互作用,对研究体系的电子结构进行了NBO分析,计算所得数据也列在表 1中,M―N键的 WBI (Wiberg)键序按照Pt(0.916) > Pd(0.631) > Cu(0.518) > Au(0.481) >Ag(0.262) > Ni(0.005)的顺序递减,N≡N 之间的WBI键序按照 Ni(2.970) > Ag(2.825) > Cu(2.760) >Au(2.704) > Pd(2.648) > Pt(2.528)的顺序递减。化学键键长与成键原子半径和成键原子之间的距离有关,WBI键序可以剔除原子半径因素对键长的影响,是描述分子中相邻原子之间的成键强度的物理量。上述计算结果表明,Pt1/POM 给出了最大的WBI(M―N)键序,和最小的WBI(N≡N)键序,说明了明显的活化氮气分子的能力。这一结果为Pt1/POM 体系潜在的固定氮气分子并使其活化提供了更有力的证据。另外对于负载过渡金属Ni的体系来说,其 WBI(M―N)值仅有 0.005,说明过渡金属与氮气之间的相互作用极弱,金属Ni与四个氧配体之间的WBI键序都达到了0.4左右,说明Ni与多酸之间相互作用较强,反而与氮气之间的相互作用很小,甚至不能达到使氮气固定在多酸催化剂上的目的,因此Ni1/POM体系不能达到活化氮气的目的。表1中给出研究体系的NBO电荷,由表中数据可知吸附的氮气分子部分的NBO电荷递增顺序为:Pd(-0.209) < Pt(-0.197) <Au(-0.121)< Ag(-0.089)< Cu(-0.083),可以看出,这些吸附后的氮气分子电荷都是负的。和中性的自由氮分子相比,氮气上的部分负电荷表明发生了电荷转移。Pt1/POM、Pd1/POM体系中氮气部分带有的负电荷达到~0.2e,表明了相对明显的电荷转移。

表1 DFT-M06L计算获得的氮气吸附能(Ead, kJ·mol-1)、M1/POM (M = Ni, Pd, Pt, Cu, Ag, Au)体系的键长(nm),金属与两个配体氧之间的键角(∠O-M-O, °)、WBI键序(WBI)和NBO电荷(Q, a.u.)Table 1 DFT-M06L-derived adsorption energy (Ead, kJ·mol-1), bond length (nm), angle of metal atom and two oxygen atoms (∠O-M-O, °), WBI bond order (WBI) and NBO partial charge (Q, a.u.) in M1/POM (M = Ni, Pd, Pt, Cu, Ag, Au).

图3 Pt1/POM的前线分子轨道分布图Fig.3 Frontier molecular orbitals of the Pt-nitrogen POM complex.

表2 M1/POM催化体系吸附氮气前后红外光谱特征峰振动频率(单位:cm-1)Table 2 Calculated and experimental vibrational frequencies (in cm-1) and the assigned bands of the series of POM complexes studied here.
前线分子轨道理论可以解释成键规律的定性理论。通过上文的NBO分析和成键性质分析可知Pt1/POM 体系相对于其它体系来说有着较为明显的活化氮气分子的能力,因此下文中主要以Pt1/POM体系为例分析它们的前线分子轨道特征。Pt1/POM体系前线分子轨道如图 3所示,可以看到,HOMO - 1轨道主要分布在负载的Pt原子上,是Pt原子的dxy轨道,表现为非键道。HOMO - 3和HOMO - 4这两个轨道是由氮气分子的π*反键轨道和过渡金属Pt原子对称匹配的dxz和dyz轨道上的电子云重叠构成的Pt-N2成键轨道。如上文所述,NBO电荷分析证明 Pt-N2成键过程发生的金属到氮气分子的电荷转移。根据 HOMO - 3和HOMO - 4轨道分析表明,Pt金属原子到氮气分子上的电荷转移,主要是由Pt金属原子的dxz、dyz轨道上的电子填充到了氮气的 π*反键轨道上引起。填入π*反键轨道的电子增长了N≡N键之间的距离,有效的活化了氮气分子。以上分析可以通过d2xyπ2xzπ2yz电子组态进行充分概括。
计算的M1/POM体系的红外光谱列于表2中。众所周知,完整的Keggin型POM是一个由氧原子连接钨原子形成的笼状结构,内部包含四面体磷酸基团,整个结构具有Td对称。实验上所测得的红外光谱数据表明,完整Keggin型POM具有四个明显的特征谱带(P-Oa,W=Ot,W-Ob-W和W-Oc-W键):P-Oa键的不对称伸缩振动出现在1080 cm-1处(Oa是四面体磷酸基团中的氧原子);W=Ot键的不对称伸缩振动出现在987 cm-1处(Ot是端氧原子);W-Ob-W 键不对称伸缩振动出现在890 cm-1(Ob代表桥氧原子);W-Oc-W 键的不对称伸缩振动出现在 802 cm-1的是(Oc代表处于转角位置的桥氧原子)30。为了验证我们计算方法的可靠性,首先采用相同的理论水平计算了完整的Keggin型POM,[PW12O40]3-,的红外光谱,计算结果也列于表2中,P-Oa(1080 cm-1)、W=Ot(976 cm-1),W-Ob-W (901 cm-1)和 WOc-W (828 cm-1),与实验测得的频率相一致(见表2),证明了计算方法的可行性。
当多酸负载过渡金属原子后,完整的 Keggin型 POM 四个明显的特征吸收峰基本没有发生变化,但是在750-770 cm-1范围内出现了新的吸收峰。如表2所示,Pd1/POM、Pt1/POM、Cu1/POM的新的特征峰分别出现在754、767、763 cm-1的位置处。对这些峰的振动模式分析表明主要是由W-Oc-W 键的伸缩振动引起。如前文所述,负载的单金属原子锚定于Keggin型POM表面的四重洞位上,主要表现为金属原子与W-Oc-W键中的Oc原子发生直接相互作用。这种成键方式影响了W-Oc-W键的伸缩振动。因此可以判断新出现的特征峰是由于负载单金属原子后从完整的Keggin型POM的W-Oc-W特征峰中分离出来的。
吸附氮气后形成的配合物的红外光谱数据也列在表2中。值得注意的是,吸附氮气分子之后,ν5(M-O)发生了移动,这可能是由于金属与氮气相互作用导致金属与多酸表面四重洞位上的氧原子之间的相互作用减弱引起的。吸附氮气分子后,在红外光谱上出现了另一个明显的红外特征吸收峰,对应于氮气分子N≡N键伸缩振动。自由氮气的红外特征吸收峰出现在2331 cm-1,N≡N键长为0.1098 nm。对双氮分子的红外光谱研究表明,当氮气分子的N≡N键伸长时,N≡N键伸缩振动频率将会减小。例如偶氮苯的红外吸收峰出现在1442 cm-1,N≡N键长为0.1255 nm;肼的红外吸收峰出现在1111 cm-1,N≡N键长为0.1460 nm。如上文所述,氮气分子的N≡N键长的顺序为:Pt(0.1139 nm) > Pd(0.1134 nm) > Cu(0.1123 nm),计算出红外光谱中N≡N键伸缩振动频率按照Pt(2194 cm-1) <Pd(2202 cm-1) < Cu(2299 cm-1)的顺序递增,与上述规律完全一致。
4 结论
采用DFT计算研究了一系列以POM为载体的SACs(M1/POM;M = Ni, Pd, Pt, Cu, Ag, Au)的分子几何构型、电子结构、红外光谱。计算结果表明这些催化剂具有潜在的活化N2分子的能力,特别是Pt1/POM体系吸附的N2分子给出了最大的N≡N键长,同时NBO分析表明Pt1/POM体系中的WBI(N≡N)的值也最小。因此,在六种研究体系中Pt1/POM的活化N2效果较好。前线分子轨道分析发现Pt1/POM中Pt金属原子的dxz、dyz轨道与氮气的π*反键轨道相互重叠使多酸载体与N2之间成键,并且金属原子中的部分电子转移到了N2的π*反键轨道导致N≡N键增长。通过对M1/POM体系的红外光谱的DFT计算结果与实验数据相比较发现,本文采用的理论方法很好的重现了过渡金属取代Keggin结构的杂多酸的红外光谱,对Pt1/POM,Pd1/POM和Cu1/POM的特征吸收峰分析表明,由于引入单原子到多酸表面产生了新的特征吸收峰。
Supporting lnformation:available free of charge νia the internet at http://www.whxb.pku.edu.cn.
(1)Burris, R. H. J. Biol. Chem. 1991, 266, 9339.
(2)Shah, V. K.; Brill, W. J. Proc. Νatl. Acad. Sci. U. S. A. 1977, 74,3249. doi: 10.1073/pnas.0507853103
(3)Chakrabarti, P.; Woo, D.;Kornuc, J. J.; Rees, D. C. Science 1992,257, 1653. doi: 10.1126/science.1529353
(4)Kirn, J.; Rees, D. C. Νature 1992, 360, 553. doi: 10.1038/360553a0
(5)Kirn, J.; Rees, D. C. Science 1992, 257, 1677.doi: 10.1126/science.1529354
(6)Chan, M. K.; Kirn, J.; Rees, D. C. Science 1993, 260, 792.doi: 10.1126/science.8484118
(7)Kirn, J.; Woo, D.; Rees, D. C. Biochemistry 1993, 32, 7104.doi: 10.1021/bi00079a006
(8)Kirn, J.; Rees, D. C. Biochemistry 1994, 33, 389.doi: 10.1021/bi00168a001
(9)Jia, H. P.; Quadrelli, E. A. Chem. Soc. Reν. 2014, 43, 547.doi: 10.1039/c3cs60206k
(10)MacKay, B. A.; Fryzuk, M. D. Chem. Reν. 2004, 104, 385.doi: 10.1021/cr020610c
(11)Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.doi: 10.1126/science.1085326
(12)Arashiba, K.; Miyake, Y.; Nishibayashi, Y. Νat. Chem. 2011,3, 120. doi: 10.1038/nchem.906
(13)Anderson, J. S.; Rittle, J.; Peters, J. C. Νature 2013, 501, 84.doi: 10.1038/nature12435
(14)Allen, A. D.; Harris, R. O.; Loescher, B. R.; Stevens, J. R.;Whiteley, R. N. Chem. Reν. 1973, 73, 11.doi: 10.1021/cr60281a002
(15)Hoffman, B. M.; Lukoyanov, D.; Yang, Z. Y.; Dean, D. R.;Seefeldt, L. C. Chem. Reν. 2014, 114, 4041.doi: 10.1021/cr400641x
(16)Qiao, B.; Wang, A.; Yang, X.; Allard, L. F.; Jiang, Z.; Cui,Y.; Liu, J.; Li, J.; Zhang, T. Νat. Chem. 2011, 3, 634.doi: 10.1038/nchem.1095
(17)Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T.Acc. Chem. Res. 2013, 46, 1740.doi: 10.1021/ar300361m.Epub 2013 Jul1
(20)Wei, H.; Liu, X. Y.; Wang, A.; Zhang, L.; Qiao, B. T.; Yang,Y. F.; Huang, Y. Q.; Miao, S.; Liu, J.; Zhang, T. Νat.Commun. 2013, 5, 5634. doi: 10.1038/ ncomms6634
(21)Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.;Chevrier, D. M.; Zhang, P.; Guo, Q.; Zang, D.; Wu, B.; Fu,G.; Zheng, N. Science 2016, 352, 797.doi: 10.1126/science.aaf5251
(22)Katsoulis, D. E. Chem. Reν. 1998, 98, 359.doi: 10.1021/cr960398a
(23)Dolbecq, A.; Dumas, E.; Mayer, C. R.; Mialane, P. Chem.Reν. 2010, 110, 6009. doi: 10.1021/cr1000578
(25)Izarova, N. V.; Pope, M. T.; Kortz, U. Angew. Chem., Int. Εd.2012, 51, 2. doi: 10.1038/srep25154
(26)Dablemont, C.; Hamaker, C. G.; Thouvenot, R.; Sojka, Z.;Che, M.; Maatta, E. A.; Proust, A. Chem. -Εur J. 2006, 12,9150. doi: 10.1002/chem.200600934
(27)Besson, C.; Musaev, D. G.; Lahootun, V.; Cao, R.;Chamoreau, L. M.; Villanneau, R.; Villain, F.; Thouvenot, R.;Geletii, Y. V.; Hill, C. L.; Proust, A. Chem. Εur. J. 2009, 15,10233. doi: 10.1002/chem.200900965
(28)Lahootun, V.; Besson, C.; Villanneau, R.; Villain, F.;Chamoreau, L. M.; Boubekeur, K.; Blanchard, S.; Thouvenot,R.; Proust, A. J. Am. Chem. Soc. 2007, 129, 7127.doi: 0.1021/ja071137t
(29)Sokolov, M. N.; Adonin, S. A.; Mainichev, D. A.; Sinkevich,P. L.; Vicent, C.; Kompankov, N. B.; Gushchin, A. L.;Nadolinny, V. A.; Fedin, V. P. Inorg. Chem. 2013, 52, 9675.doi: 10.1021/ic401492q
(30)Liu, C. G.; Liu, S.; Zheng, T. Inorg. Chem. 2015, 54, 7929.doi: 10.1021/acs.inorgchem.5b01002
(31)Coperet, C.; Comas-Vives, A.; Conley, M. P.; Estes, D. P.;Fedorov, A.; Mougel, V.; Nagae, H.; Nunez-Zarur, F.;Zhizhko, P. A. Chem. Reν. 2016, 116, 323.doi: 10.1002/chin.201615211
(32)Zhang, B.; Asakura, H.; Zhang, J.; Zhang, J.; De, S.; Yan, N.Angew. Chem. Int. Εd. 2016, 55, 8319-8323.doi: 10.1002/anie.201602802
(33)Zhang, B.; Asakura, H.; Yan, N. Ind. Εng. Chem. Res. 2017,56, 3578. doi: 10.1021/acs.iecr.7b00376
(34)Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101.doi: 10.1063/1.2370993
(35)Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270.doi: 10.1063/1.448799
(36)Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299.doi: 10.1063/1.448975
(37)Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Reν. 2005, 105,2999. doi: 10.1021/cr9904009
(38)Glendening, A. E.; Carpenter, J. E.; Weinhold, F. ΝBO Version 3.1.
(39)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
(40)Zhang, B.; Asakura, H.; Zhang, J.; Zhang, I.; De, S.; Yan, N.Angew. Chem. Int. Εd. 2016, 55, 1.doi: 10.1002/anie.201602801
