采用全外显子测序分析技术诊断X连锁慢性肉芽肿病一家系
2018-04-03许珊珊李奇渊吴谨准陈国兵朱碧溱谷为岳
许珊珊, 李奇渊,2, 吴谨准, 陈国兵, 朱碧溱, 谷为岳
慢性肉芽肿病(chronic granulomatous disease,CGD)是一种罕见的原发性免疫缺陷病,是因编码NADPH氧化酶的基因缺陷引起。CGD的遗传方式有X连锁隐性遗传(XR-CGD)及常染色体隐性遗传(AR-CGD)等,其中X连锁隐性遗传更为常见[1]。XR-CGD是由编码NADPH氧化酶的蛋白亚基gp91phox的CYBB基因突变引起[2-4]。2015年7月15日,笔者医院收治1例临床表现为反复感染,入院24 h内迅速死亡的患者,采用二代测序方法进行全外显子测序分析,对患者及家属进行Sanger测序验证,发现该患者携带CYBB基因上一个错义突变(c.949T>A, p.M312K),为半合子突变,确诊为XR-CGD,报道如下。
1 病例介绍
1.1临床资料 患者,男性,1岁3月,以“发热10余天”为主诉入院。患者入院前10余天无明显诱因出现发热,经退热抗感染治疗无效,伴精神差、纳差。出生后正常接种卡介苗。患儿2月大时出现左腋下淋巴结肿大,生后多次反复发热,反复发生肛周脓肿及头皮疖肿。入院查体:体温36.0 ℃,脉搏116 min-1,呼吸32 min-1,血压120/68 mmHg(1 mmHg=133.3 Pa),动脉血氧饱和度(arterial oxygen saturation,SaO2)96%。面色苍白,精神差,全身皮肤散在陈旧性皮疹,部分有结痂,左腋下可及一肿大淋巴结,大小约4 cm×5 cm,边界清楚,无触痛,余浅表淋巴结未触及肿大。咽充血,咽后壁可见一溃疡,未见分泌物。颈软。双肺呼吸音粗,未闻及干、湿啰音。心音有力,律齐,心前区闻及Ⅱ/Ⅵ收缩期杂音。腹稍胀,肝肋下4~5 cm可及肿大,脾肋下未触及肿大,肠鸣音4 min-1,肛周及外阴皮肤潮红,未见破损。病理征阴性,CRT 1 s。入院后患儿生命征不稳定,出现休克症状,予扩容补液、抗感染、呼吸机辅助通气,予血管活性药物、输注血液制品、镇静等对症处理。后患儿出现心跳骤停,经抢救无效于2015-07-16死亡。
1.2二代测序 在知情同意的前提下,采集患者及其家属全血各2 mL(EDTA抗凝血),使用血液基因组中量提取试剂盒(Blood Gen Midi Kit,中国CWBIO公司)提取全基因组DNA。参考文献及OMIM数据库、NCBI数据库的信息,将人类已知的全部(约2万个)基因的基因组外显子区域定制罗氏Nimble Gen捕获探针,进行全外显子捕获。合格的基因组DNA被Cavoris仪随机打断至200 bp左右,片段化DNA在Klenow Fragment、T4 DNA polymerase和T4PNK的作用下进行末端补平修复。在聚合酶体系下,使上一步得到的修复产物在3′末端加上A碱基,最后在两端加上接头。加上接头后的连接产物经4~6轮LM-PCR扩增,将文库与探针杂交60~68 h,使用链霉素磁珠进行洗脱,洗脱产物经10轮LM-PCR扩增。PCR产物在Illumina hiseq2500平台标准化上机测序,测序数据利用Illumina的软件进行分析处理。参考序列为hg19基因组。根据测序深度及突变质量,对检测到的变异进行过滤筛选,依据1000Genomes Project,Exome Variant Server (EVS)及Exome Aggregation Consortium (ExAC)数据库,滤除人群中突变频率>1%的变异,去除内含子变异、同义变异(除外位于剪切位点的变异)等,筛选出可能致病的变异,同时使用SIFT等软件对筛选出的突变对蛋白功能的影响进行预测。结合患儿的临床表现及遗传方式,得到可能致病的突变。
1.3一代测序(Sanger法)验证 根据CYBB基因所验证位点序列设计引物,采用PCR方法进行扩增。PCR扩增产物用ABI 3730XL测序仪测序,基因序列分析采用DNASTAR软件进行序列分析和比对。
1.4结果
1.4.1实验室检查结果 该患儿血液免疫功能相关检查基本正常(表1)。
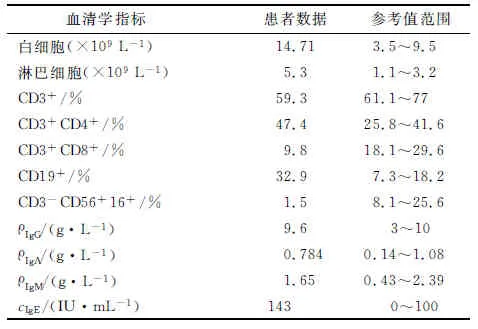
表1 患儿血液实验室检查相关结果
1.4.2测序结果 目标区域平均测序深度为66.08,目标区域覆盖度99.7%,89.76%目标区域>10×覆盖度,78.11%目标区域>20×覆盖度。
二代测序方法进行全外显子测序分析,发现患者的CYBB基因第9外显子存在半合子突变,进一步对患者及其家属用Sanger测序法验证该位点的突变情况,证明患者CYBB基因携带一个已知错义突变(c.949T>A, p.M312K),来源于母亲及外婆(图1),而其小姨并未携带该突变(参考序列NM-000397.3)。全外显子测序结果还显示,与免疫缺陷性疾病有关的3个基因存在碱基变异(表2),但后续通过遗传方式及与临床表型的关联程度排除了该3个变异的致病性。
2 讨 论
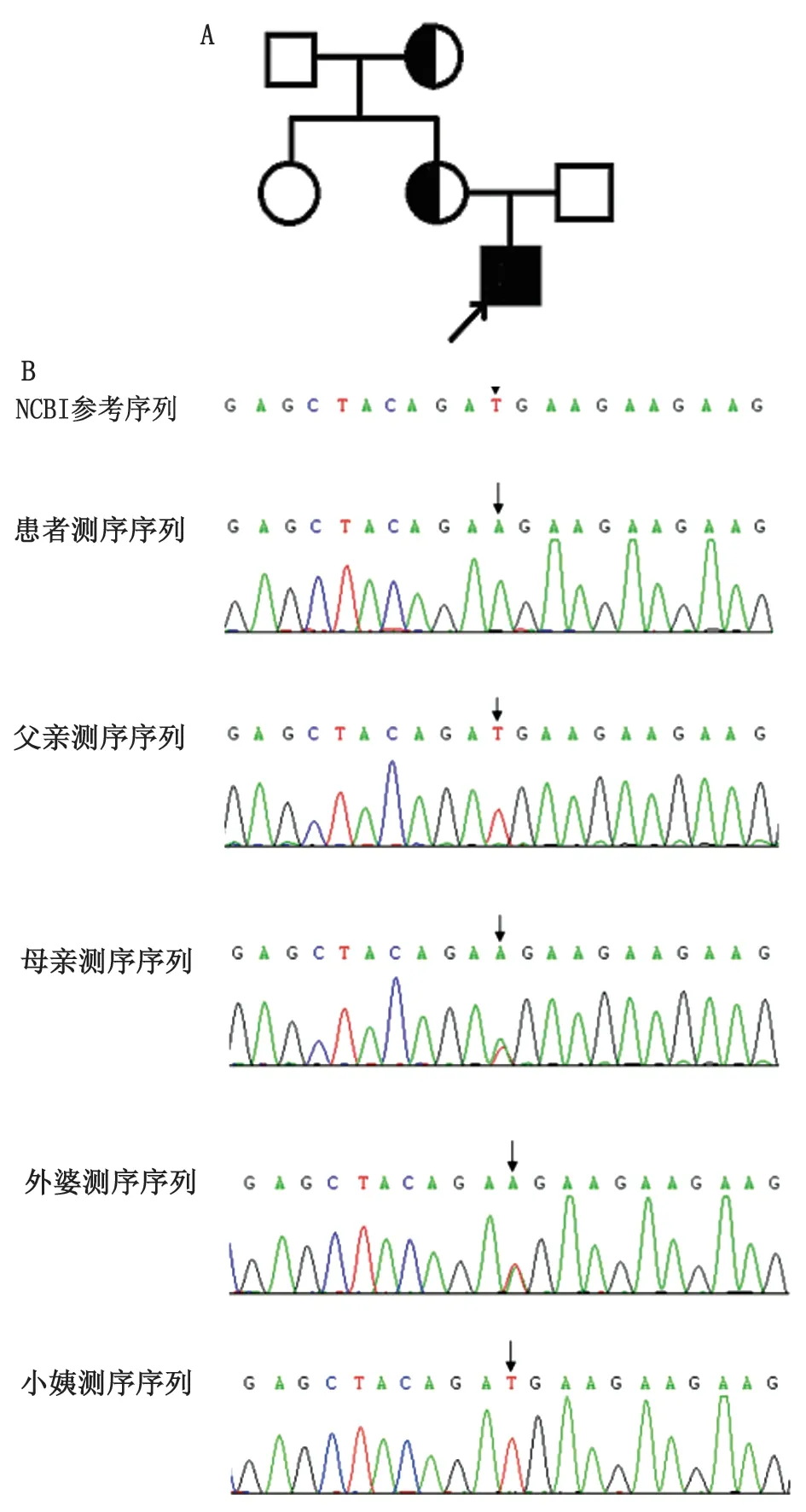
A:患者家系图,其母亲、外祖母均为该突变的携带者;B:患者及其父母、外婆、小姨Sanger测序验证CYBB基因c.949碱基改变. 参考序列NM-000397.3.图1 患者家系图及测序图谱Fig 1 Pedigree and Sanger sequencing resultsof the family
Tab2Summary of variations in other genes related with immunodeficiency disease revealed by whole exome sequencing in this patient
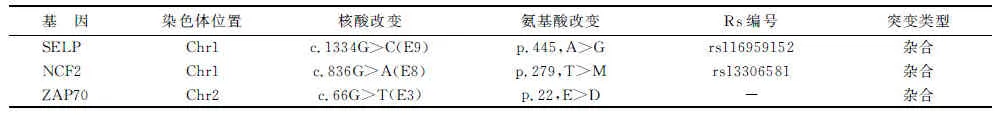
基因染色体位置核酸改变氨基酸改变Rs编号突变类型SELPChr1c.1334G>C(E9)p.445,A>Grs116959152杂合NCF2Chr1c.836G>A(E8)p.279,T>Mrs13306581杂合ZAP70Chr2c.66G>T(E3)p.22,E>D-杂合
CGD是一种罕见的原发性免疫缺陷病,是由吞噬细胞功能缺陷引起的,发病率大约为1/250 000~1/200 000。其主要临床表现是严重的反复的细菌及真菌感染,以及因反复炎症造成的胃肠道、泌尿生殖道等器官的肉芽肿增生。临床诊断主要依靠典型的临床症状、硝基四唑氮蓝(NBT)试验及中性粒细胞呼吸爆发试验等[5-6],确诊仍需依靠基因检测。本研究中,患者入院时已处于感染导致脓毒血症的状态,后迅速出现休克,于入院第2天抢救无效死亡。结合患者既往多次感染病史,考虑其患有某种免疫缺陷病,患者死亡时免疫功能相关检查(体液免疫及细胞免疫功能)尚未回报,因此在征得患者家属同意的情况下,及时外送基因检测公司进行全外显子测序,最终证实了XR-CGD的诊断。回顾性分析该患者的临床表现,与CGD相符。其主要表现为反复感染,感染部位累及皮肤(头皮、肛周)及肺,同时还有卡介苗接种同侧腋窝淋巴结肿大。实验室检查提示,细胞及体液免疫功能正常。因此,对于临床上有患者出现反复感染,怀疑免疫缺陷,但常规细胞免疫及体液免疫功能却正常的患者,应考虑CGD可能。
CGD是因编码NADPH氧化酶的基因缺陷引起。NADPH氧化酶由5个蛋白亚单位组成:gp91phox,p22phox,p47phox,p67phox,p40phox。其中XR-CGD是最常见的类型,约占CGD病例的60%,是由编码gp91phox蛋白的CYBB基因突变引起[3-4]。CYBB基因位于Xp21.1,该基因长30 kb,包含13个外显子。据HGMD网站报道,截止至2015年12月已发现CYBB基因突变700余种,最常见的突变类型是错义突变。不同的突变引起吞噬细胞gp91phox蛋白功能表达的不同,从而引起氧化酶功能障碍[2]。
本例患者携带的突变为c.949T>A,引起第312位蛋白质由蛋氨酸变为赖氨酸。该突变由Lee于2008年报道[7],Lee的研究中携带该突变的患者的临床表现为肺脓肿、肛周脓肿及淋巴结炎,无结核及卡介苗接种并发症。本研究中,患者接种卡介苗的同侧腋窝淋巴结出现明显肿大,余淋巴结未见肿大,但因未行淋巴结活检,未能推测其是否有淋巴结结核或卡介苗感染。Lee等报道,17例患者中有14例患有结核或卡介苗相关并发症,患病率较高[7]。在我国,卡介苗属于计划免疫的部分,一般在生后3 d内接种,所以对于有免疫缺陷病家族史的患儿,避免接种卡介苗可减少结核或卡介苗感染的发病率。目前美国已采用TREC试验对重症联合免疫缺陷病(severe combined immunodeficiency, SCID)进行筛查[8],期待未来能针对CGD、SCID等免疫缺陷病开展新生儿筛查。
Roos等还报道过1例CYBB基因第312位蛋白质改变的病例[2]。该患者的947位碱基T被替换为G,从而导致蛋氨酸变为精氨酸,进行蛋白表达及酶学测定后,判定其为x91+型,即gp91phox蛋白表达正常,NADPH氧化酶活性缺失[4]。本病例与该病例相比,同为第949位碱基发生改变,导致312位蛋白质变为精氨酸或赖氨酸,二者均有带正电荷的侧链,同为碱性氨基酸。所以推测c.949T>A(p.M312K)可能也为x91+型,可引起酶活性基本缺失。
近几年,随着二代测序技术的发展,费用降低,测序速度加快,已成为罕见单基因遗传病诊断的重要方法之一。通过二代测序技术,可以快速地对目的基因组合或全外显子进行测序,使得对罕见病、疑难病的诊断变得更加容易[9]。如本例患者,病情进展迅速,入院1 d内即死亡,临床实验室检查尚来不及完善,而通过二代测序技术进行全外显子测序分析,明确诊断,并可对家属提供遗传咨询。由于同时对全外显子进行测序,测序结果可能会提示多个基因携带变异。如本研究全外显子测序结果显示了另外3个与免疫缺陷性疾病有关的基因上存在碱基变异。与SELP基因及NCF2基因相关的疾病为遗传性IgE反应过敏症及遗传CGD细胞色素B阳性2型,均为常染色体显性遗传,但在此2个基因上发现的变异均为NCBI dbSNP数据库中记载的已知SNP位点,其临床意义为良性。因ZAP70突变引起的选择性T细胞缺陷病为常染色体隐性遗传病,该患者携带ZAP70杂合突变,故不会致病。所以全外显子测序检测出的这3个基因变异均无临床意义。这就需要临床医师进行鉴别分析,过滤或剔除无用信息,找到真正的致病突变。
综上所述,对于临床症状不典型,或因不可抗拒原因(如患者死亡)而造成临床资料不完善,无法进行临床诊断时,采用二代测序技术进行目的基因组合或全外显子测序,可以帮助医师明确诊断,并对患者家庭提供可靠的遗传咨询。
[1] Segal B M, Leto T L, Gallin J I,etal. Genetic, biochemical and clinical features of chronic granulomatous disease[J].Medicine(Baltimore), 2000,79:170-200.
[2] Roos D, De Boer M, Kuribayashi F,etal. Mutations in the X-linked autosomal recessive forms of chronic granulomatous disease[J].Blood, 1996,87:1663-1681.
[3] Rae J, Newburger P E, Dinauer M C,etal. X-linked chronicgranulomatous disease: Mutations in the CYBB gene encoding the gp91-phox-component of therespiratory burst oxidase[J].AmJHumGenet, 1998,62:1320-1331.
[4] Roos D, Kuhns D B, Maddalena A,etal. Hematologically important mutations: X-linked chronic granulomatous disease (third update)[J].BloodCellsMolDis, 2010,45(3):246-265.
[5] Ochs H D, Igo R P. The NBT slide test: a simple screening method for detecting chronic granulomatous disease and femalecarriers[J].JPediatr, 1973,83(1):77-82.
[6] Alvarez-Larran A, Toll T, Rives S,etal. Assessment ofneutrophil activation in whole blood by flow cytometry [J].ClinLabHaematol, 2005,27(1):41-46.
[7] Lee P P, Chan K W, Jiang L,etal. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease[J].PediatrInfectDisJ, 2008,27(3):224-230.
[8] Chinn I K, Shearer W T. Severe combined immunodeficiency disorders[J].ImmunolAllergyClinNorthAm, 2015,35(4):671-694.
[9] Yu Y, Wu B L, Wu J,etal. Exome and whole-genome sequencing as clinical tests: a transformative practice in molecular diagnostics[J].ClinChem, 2012,58(11):1507-1509.
