偏向性配体
——阿片类镇痛药设计新思路
2018-04-03苏瑞斌
孙 毅 谭 博 苏瑞斌
抗毒药物与毒理学国家重点实验室,神经精神药理学北京市重点实验室,军事科学院军事医学研究院毒物药物研究所,北京,100850,中国
吗啡等阿片类药物是目前临床上最常使用的一类镇痛药[1],此类药物在治疗疼痛的同时也伴有耐受、胃肠功能紊乱、呼吸抑制等副作用。除此之外,阿片类物质在使用过程中诱导欣快感和易成瘾的特点使得该类药物在毒品市场上大量非法流通,给无数家庭和社会带来巨大危害[2]。在这些副作用中,呼吸抑制是限制阿片类药物用药安全性的主要原因[3]。过去几十年里,因阿片类药物滥用引起呼吸抑制而死亡的人数一直在上升,且预计短期内人数仍将居高不下[4-5]。因此,自19世纪以来,人们就一直致力于设计、优化阿片类药物结构,希望得到副作用更低而安全性更高的强效镇痛药。
阿片的应用已有几千年历史,但人们对其主要作用靶标阿片受体(opioid receptor,OR)的认识却相对滞后。阿片受体属于G蛋白偶联受体(G protein-coupled receptor,GPCR),主要可分为μ、δ、κ阿片受体和痛敏肽受体(孤儿受体)四种亚型[6]。最初,人们寄希望于受体亚型特异性配体以规避阿片类药物常见的副作用,在这一理念的指导下,人们合成了新型化合物美沙酮、芬太尼等,也陆续发现了各个亚型的特异性内源阿片肽[7-8],但其副作用的问题依然没有得到解决。至90年代,随着基因克隆技术的发展,科学家们发现吗啡所有可观察到的效应在μ阿片受体(μ-opioid receptor,μOR)敲除的模式生物上都消失了,这提示无论是镇痛、镇静效应还是呼吸抑制、胃肠功能紊乱、成瘾等副作用都主要通过μOR[9],受体亚型选择性理论难以解释这一现象。
近年研究发现,GPCR激活后,在其下游除了经典的G蛋白依赖型信号通路外,还存在另一条由arrestin介导的平行的信号通路,不同的配体可以不同程度地激活这两条通路,甚至只激活其中一条。因此,最终表现在整体动物上的药理效应也不同(Fig.1)[10]。有关μOR的研究进一步显示,阿片类药物产生的镇痛、镇静、欣快感以及躯体依赖主要由G蛋白依赖型信号通路介导,而其致耐受、胃肠功能紊乱、呼吸抑制等副作用则由β-arrestin依赖型信号通路介导[11]。如果某个配体偏向性激活G蛋白信号通路,那么该配体可能在保持镇痛、镇静活性的同时,副作用相对于经典的阿片类药物大大降低。可以说,arrestin依赖型信号通路的发现以及偏向性配体理论的提出给设计新型副作用降低的阿片类镇痛药提供了思路。
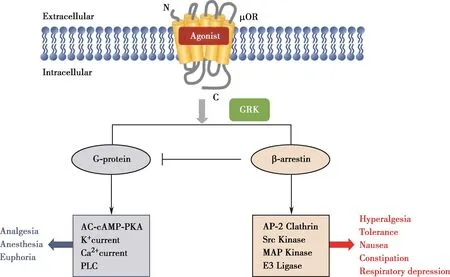
Fig.1 G protein-dependent and β-arrestin-dependent signaling through μOR
1 β-arrestin在阿片受体下游信号通路中的功能
Arrestin是GPCR下游信号通路的重要负性调节因子。目前在哺乳动物体内发现的arrestin主要有四种,分别被命名为arrestin1~4,其中arrestin1和arrestin4存在于视网膜干体和锥体细胞上,主要结合视紫红质蛋白;而arrestin2和arrestin3广泛分布于全身,也分别被称为β-arrestin1和β-arrestin2[12]。Oakley根据受体与arrestin相互作用的特点将GPCR分为A、B两类,其中A类GPCR仅结合β-arrestin,且与β-arrestin2结合的紧密程度要大于β-arrestin1;B类GPCR与β-arrestin1和β-arrestin2的亲和力相近,同时也可以与arrestin1和arrestin4结合。除此之外,A类GPCR与arrestin的结合是瞬时的,而B类GPCR与arrestin的结合则较为稳定,在这一分类下,阿片受体属于A类GPCR[13]。
1.1 β-arrestin是GPCR下游G蛋白依赖型信号通路的抑制蛋白
上世纪90年代以前,人们对μ阿片受体下游G蛋白介导的信号通路就有所了解。与经典的GPCR信号通路一致,当激动剂作用于μ阿片受体后,受体偶联G蛋白,G蛋白随即被激活分解为Gα亚基和Gβγ亚基复合物,其后这两种信号分子继续作用于腺苷酸环化酶(adenylate cyclase,AC)、磷 脂 酶 C(phospholipase C,PLC)和K+、Ca2+离子通道,从而引起下游信号分子的变化[14]。Arrestin最初被认为是终结这一反应的负性调节因子,当GPCR被G蛋白激酶(G-protein-coupled receptor kinase,GRK)磷酸化之后,受体C末端开始招募arrestin,arrestin与受体结合占据的位置在空间上阻碍了G蛋白与受体的相互作用,因此G蛋白依赖型信号通路被阻断,这可能是受体脱敏的主要原因[15]。
1.2 β-arrestin作为衔接蛋白参与受体内吞和泛素化
此后,一系列研究发现β-arrestin在许多GPCR(包括μOR在内)的内吞和泛素化中起到了重要作用[16-17]。
β-arrestin被受体招募后,可作为衔接蛋白连接受体至网格蛋白包被小窝(clathrin-coated pits)中[18]。在这一过程中,β-arrestin至少直接与两个内吞相关分子作用,一个就是网格蛋白(clathrin),另一个是异四聚体网格衔接蛋白AP-2。当受体C末端被GRK磷酸化之后,β-arrestin结合受体,之后招募网格蛋白和AP-2,致使细胞膜上的受体-β-arrestin复合物进入小窝中。然后,受体内吞至酸性核内体,在酸性核内体中的受体一部分去磷酸化后回收至细胞膜,而另一部分被降解,即受体下调[19]。因此,β-arrestin作为衔接蛋白参与了受体内吞的过程。
除此之外,β-arrestin还作为E3连接酶衔接蛋白参与受体泛素化,这一现象最初是在β2肾上腺素受体中发现[20]。进一步的研究表明β-arrestin是作为E3泛素连接酶 NEDD4(neural precursor cell expressed,developmentally downregulated 4,NEDD4)的支架蛋白促使受体泛素化[21]。
1.3 β-arrestin作为GPCR下游信号传导器
以往研究结果使人们对β-arrestin的认识停留在G蛋白信号通路的负性调节因子上,近几年的最新研究成果显示β-arrestin还可以作为GPCR的信号传导器,独立于G蛋白介导下游信号通路的传递。
β-arrestin被发现可以作为信号传导器最初的证据是其可以作为支架蛋白将酪氨酸激酶c-Src(Src kinase)家族招募至GPCR上,而Src激酶家族不仅参与细胞的增殖与分化,也是丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/细胞外信号调 节 激 酶(extracellular signal-regulated kinase,ERK)信号通路上的重要组成部分[22]。这一发现让人们意识到GPCR下游不仅只有G蛋白介导的信号通路,还存在一条与G蛋白通路平行的β-arrestin依赖型信号通路[23]。此后,结果表明β-arrestin作为支架蛋白参与两条信号通路,一条是上述MAPK/ERK通路,另一条是c-JUN氨基末端激酶3(c-JUN N-terminal kinase type 3,JNK3)通路[24-25]。β-arrestin作为支架蛋白组装了通路中相互作用的蛋白,确保信号正确、特异地传递[26]。目前,对GPCR下游β-arrestin依赖型信号通路还知之甚少,唯一能确定的就是β-arrestin的功能远远超出当前的认知。
2 偏向性配体理论的提出与发展
以往,人们曾认为配体发挥不同的药理效应是因为其作用的受体不同,而受体只有“开”或“关”两种状态分别对应“有”或“无”两种生理反应[26]。80年代末期,人们开始意识到配体发挥药理效应并不完全取决于其作用的受体,而是和受体下游的信号分子相关[27]。90年代,研究证明了在相同的细胞环境下,配体作用于同一受体,并不是平均地激活下游每一条通路,而是对不同信号通路的激动活性不同[28]。因此,配体与受体的相互作用就不能用简单的“开”“关”模式形容。随着GPCR结构解析研究的进展,人们认为受体的激活构象是动态的、可变的,与不同配体结合,受体呈现出不同的激动态结构,从而不同程度地招募G蛋白和β-arrestin,触发不同的信号通路,最终引起千差万别的效应[29-30]。之后,人们发现某些配体偏向性地激活受体后某条信号通路,而较少或不激活受体后的其它通路,由此 “偏向性配体”的概念被逐渐提出,即配体并不是平均地激活GPCR下游的每一条信号通路,而是可能选择性、偏向性地激活某些信号通路,这种现象也称为“偏向性激活”[31]。
β-arrestin依赖型信号通路发现后,人们又很快发现G蛋白依赖型信号通路与β-arrestin依赖型信号通路对激动剂诱导的药理作用的贡献是不同的。有趣的是,对于μOR来说,G蛋白依赖型信号通路介导镇痛、镇静效应,而β-arrestin依赖型信号通路则主要介导其他药理效应[32]。
近年来许多研究采用基因敲除的方法探讨了β-arrestin与阿片类镇痛药的药理效应之间的关系。β-arrestin1和β-arrestin2完全敲除后有胚胎致死性,敲除小鼠无法成活,而单独敲除β-arrestin1或β-arrestin2,则可以得到表型正常的小鼠,提示β-arrestin是小鼠生长发育过程中的必要蛋白,而β-arrestin1和β-arrestin2之间的功能可能相互替代[33-35]。对于β-arrestin1敲除小鼠,单剂量的吗啡镇痛效应和野生型小鼠类似[36];而在β-arrestin2敲除小鼠中,吗啡表现出了更强的镇痛活性,受体脱敏也明显减弱[33-35,37]。采用 RNA 干扰敲低β-arrestin的小鼠和大鼠中也观察到了类似的现象[38]。除镇痛效应外,还有研究表明吗啡引起的便秘和呼吸抑制在β-arrestin2敲除小鼠中相对于野生型小鼠也明显减弱。然而,并不是所有的阿片类药物在β-arrestin2敲除小鼠上都表现出镇痛效应增强,美沙酮、芬太尼和埃托啡在β-arrestin2敲除小鼠上的镇痛效应与野生型小鼠类似[36-37]。最新研究对芬太尼等阿片激动剂在分子水平进行偏倚系数测定,同时在整体动物水平对这些药物的镇痛效应和呼吸抑制副反应进行测定,结果显示配体激活信号通路的偏倚和配体安全窗口大小有关。当配体偏向性激活β-arrestin依赖型信号通路时,配体安全窗口较窄[39]。
以上实验结果提示阿片类药物引起的便秘、呼吸抑制等副反应可能主要由β-arrestin2介导。如果能够设计出只激活G蛋白依赖型信号通路的偏向性配体,那么该配体可能在保持其镇痛、镇静活性的同时,其他药理效应(副作用)相对于经典的阿片类药物会大大降低。
目前研究认为配体与受体结合,不同结构的配体使受体处在不同的稳定构象中,导致下游信号传递分子结合或激活的差异,从而产生不同的信号,最终体现在动物水平上的药理效应也不一样[30]。部分受体结构解析研究提示受体中的某些基元(比如NPxxY)可能和β-arrestin的结合有关[40],而某些基元(比如DRY和PIF)可能和G蛋白信号通路相关[11]。然而,受体介导偏向性信号通路的机制目前还不清楚。
3 基于偏向性配体理论设计的药物
偏向性配体理论提示μ阿片受体下游G蛋白依赖型信号通路的偏向性激动剂可能在保持镇痛活性的同时,较少引起便秘、呼吸抑制等副作用。科学家们基于这一理论广泛设计筛选化合物,得到了一系列潜在的偏向性配体,其中较为成功的化合物主要有TRV130和PZM21。
3.1 TRV130
为了找到μ阿片受体G蛋白通路偏向性配体,科学家对现有化学库中的大量化合物分别进行G蛋白信号通路和β-arrestin信号通路活性的测定,初筛出一系列表现出G蛋白信号通路偏向性的化合物。在比较、分析这些化合物构效关系的基础上,从效能、偏向性、选择性等方面对化合物结构进行了不断优化,最终得到了TRV130。在细胞实验上,研究者发现TRV130相对吗啡而言是β-arrestin偏向性激动剂,在小鼠和大鼠实验上,TRV130较同等剂量下的吗啡显示出相似的镇痛效应,但胃肠功能紊乱和呼吸抑制等副作用则明显减弱[41]。2016年美国食品药品监督管理局(Food and Drug Administration)批准TRV130进入三期临床研究,结果显示Oliceridine(TRV130商品名)起效比吗啡快,用药后呕吐现象减少,但只有最低剂量的Oliceridine所致的呼吸抑制作用比吗啡更轻微,同时只有最高剂量的Oliceridine的镇痛作用比吗啡更强[42]。
3.2 PZM21
相对于TRV130从大量化合物的构效关系研究入手,PZM21的设计策略则是基于对μOR的结构分析。在已解析μOR结构的基础上,将300万个化合物与μOR的正性结合口袋进行对接研究,从亲和力排在前2 500名的化合物中找出23个结构新颖且与受体结合关键氨基酸位点D1473.32作用的化合物。将这23个化合物进行活性和偏向性检测,从中选出了一个强烈激活G蛋白依赖型信号通路而较少引起β-arrestin募集的化合物,对这个化合物进行结构优化后,最终得到了PZM21。相对于TRV130,PZM21在细胞水平上对μOR表现出了更高的亚型选择性。动物实验上,与等剂量的吗啡相比,PZM21显示出更强的镇痛活性,而其诱导呼吸抑制和便秘的作用明显降低[43]。以上结果均提示PZM21可能会成为一个非常值得期待的新型镇痛药。
4 展望
历史上,人们为了解决阿片类镇痛药的副作用问题,做了很多尝试,比如设计阿片受体亚型选择性激动剂[8]、设计正向变构调节剂[44]等,但到目前为止尚不能有效地解决其多种副作用。β-arrestin依赖型信号通路的发现以及偏向性配体理论的提出给设计新型副作用更低的强效阿片类镇痛药提供了新的思路。这一新思路引起了广泛的关注,但最终转化成临床可用的副作用降低的镇痛药还面临许多挑战。一方面,尽管我们从动物敲除实验了解到G蛋白依赖型信号通路和β-arrestin依赖型信号通路分别介导性质不同的药理效应,但G蛋白或β-arrestin下游的具体信号通路与最终药理效应的对应关系目前还不清楚。另一方面,我们对受体和配体相互作用的结构机制还不完全了解,配体具有什么样的特点能被受体识别并偏向性激活某一条信号通路?配体微小结构的差异如何引起受体结构的变化,最终引起不同信号通路的传递?这些问题还没有答案。除了以上这两方面,如何测定、评估配体的偏向性目前仍存在争议[45]。值得期待的是,受体后信号通路的偏倚现象普遍存在于所有的GPCR中,偏向性配体理论不仅有可能解决阿片类药物副作用的问题,还可能为靶向其他GPCR的药物设计提供思路,科研工作者可以根据实际需求自主设计药物,选择性调控G蛋白依赖型信号通路或β-arrestin依赖型信号通路。总之,虽然偏向性配体药物走向市场还面临一些挑战,但随着人们对GPCR与配体相互作用以及受体后信号通路的不断深入了解,偏向性配体在未来会广泛应用于临床。
[1] Thompson G L,Kelly E,Christopoulos A,et al. Novel GPCR paradigms at the mu-opioid receptor[J]. J British Pharmacology,2015,172(2):287-296.
[2] Andrew Kolodny,David T Courtwright,Courtwright S Hwang,et al. The prescription opioid and heroin crisis:a public health approach to an epidemic of addiction[J].Annual Review of Public Health,2015,36:559-574.
[3] Imam M Z,Kuo A,Ghassabian S,et al. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression[J]. Neuropharmacology,2017,131:238-255.
[4] Richard G Frank,Harold A Pollack. Addressing the fentanyl threat to public health[J]. J New England Medicine,2017,376(7):605-607.
[5] Nora D Volkow,A Thomas McLellan. Opioid abuse in chronic pain--misconceptions and mitigation strategies[J].J New England Medicine,2016,374(13):1253-1263.
[6] Gavril W Pasternak. Opioids and their receptors:Are we there yet? [J]. Neuropharmacology,2014,76 Pt B:198-203.
[7] Hughes J,Smith T W,Kosterlitz H W,et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity[J]. Nature,1975,258(5536):577-580.
[8] Gilbert P E,Martin W R:The effects of morphine and nalorphine-like drugs in the nondependent,morphinedependent and cyclazocine-dependent chronic spinal dog[J]. J Pharma cology and Experimental Therapeutics,1976,198(1):66-82.
[9] Hans W D Matthes,Rafael Maldonado,Frederic Simonin,et al. Loss of morphine-induced analgesia,reward effect and withdrawal symptoms in mice lacking the mu-opioidreceptor gene[J]. Nature,1996,383(6603):819-823.
[10] Jonathan D Violin,Aimee L Crombie,David G Soergel,et al. Biased ligands at G-protein-coupled receptors:promise and progress[J]. Trends in Pharmacological Sciences,2014,35(7):308-316.
[11] Huang Wei-jiao,Aashish Manglik,A J Venkatakrishnan,et al. Structural insights into micro-opioid receptor activation[J]. Nature,2015,524(7565):315-321.
[12] Attramadal H,Arriza J L,Aoki C,et al. Beta-arrestin2,a novel member of the arrestin/beta-arrestin gene family[J].J Biological Chemistry,1992,267(25):17882-17890.
[13] Oakley R H,Laporte S A,Holt J A,et al. Differential affinities of visual arrestin,beta arrestin1,and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors[J]. J Biological Chemistry,2000,275(22):17201-17210.
[14] Yuri K Peterson,Louis M Luttrell. The diverse roles of arrestin scaffolds in g protein-coupled receptor signaling[J]. Pharmacological Reviews,2017,69(3):256-297.
[15] Louis M Luttrell,Robert J Lefkowitz. The role of betaarrestins in the termination and transduction of G-proteincoupled receptor signals[J]. J Cell Science,2002,115(Pt 3):455-465.
[16] Kirsten M Raehal,Laura M Bohn. beta-arrestins:regulatory role and therapeutic potential in opioid and cannabinoid receptor-mediated analgesia[J]. Handbook of Experimental Pharmacology,2014,219:427-443.
[17] Reddy Peera Kommaddi,Sudha K Shenoy. Arrestins and protein ubiquitination[J]. Progress in Molecular Biology and Translational Science,2013,118:175-204.
[18] McDonald P H,Lefkowitz R J. Beta-Arrestins:new roles in regulating heptahelical receptors’ functions[J]. Cellular Signalling,2001,13(10):683-689.
[19] Laporte S A,Oakley R H,Zhang J,et al. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis[J]. Proceedings of the National Academy of Sciences of the United States of America,1999,96(7):3712-3717.
[20] Sudha K Shenoy. Seven-transmembrane receptors and ubiquitination[J]. Circulation Research,2007,100(8):1142-1154.
[21] Sudha K Shenoy,Xiao Kun-hong,Vidya Venkataramanan,et al. Nedd4 mediates agonist-dependent ubiquitination,lysosomal targeting,and degradation of the beta2-adrenergic receptor[J]. J Biological Chemistry,2008,283(32):22166-22176.
[22] Luttrell L M,Ferguson S S,Daaka Y,et al. Beta-arrestindependent formation of beta2 adrenergic receptor-Src protein kinase complexes[J]. Science (New York,NY),1999,283(5402):655-661.
[23] Erik G Strungs,Louis M Luttrell. Arrestin-dependent activation of ERK and Src family kinases[J]. Handbook of Experimental Pharmacology,2014,219:225-257.
[24] Gong Kai-zheng,Li Zi-jian,Xu Ming,et al. A novel protein kinase A-independent,beta-arrestin-1-dependent signaling pathway for p38 mitogen-activated protein kinase activation by beta2-adrenergic receptors[J]. J Biological Chemistry,2008,283(43):29028-29036.
[25] Patricia H McDonald,Chi-Wing Chow,William E Miller,et al. A receptor-regulated MAPK scaffold for the activation of JNK3[J]. Science (New York,NY),2000,290(5496):1574-1577.
[26] Pierce K L,Lefkowitz R J:Classical and new roles of betaarrestins in the regulation of G-protein-coupled receptors[J]. Nature Reviews Neuroscience,2001,2(10):727-733.
[27] Samama P,Cotecchia S,Costa T,et al. A mutation-induced activated state of the beta 2-adrenergic receptor[J].Extending the ternary complex model. The Biological Chemistry,1993,268(7):4625-4636.
[28] Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals[J]. Trends in Pharmacological Sciences,1995,16(7):232-238.
[29] Dean P Staus,Strachan R T,Manglik A,et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation[J]. Nature,2016,535(7612):448-452.
[30] Manglik A,Kruse A C. Structural basis for g proteincoupled receptor activation[J]. 2017,56(42):5628-5634.
[31] Terry P Kenakin,Arthur Christopoulos. Signalling bias in new drug discovery:detection,quantification and therapeutic impact[J]. Nature Reviews Drug discovery,2013,12(3):205-216.
[32] Kelly E. Efficacy and ligand bias at the mu-opioid receptor[J]. British J Pharmacology,2013,169(7):1430-1446.
[33] Laura M Bohn,Robert J Lefkowitz,Raul R Gainetdinov,et al. Enhanced morphine analgesia in mice lacking betaarrestin 2[J]. Science (New York,NY),1999,286(5449):2495-2498.
[34] Laura M Bohn,Raul R Gainetdinov,Lin Fang-Tsyr,et al. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence[J].Nature,2000,408(6813):720-723.
[35] Laura M Bohn,Robert J Lefkowitz,Marc G Caron.Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice[J].The J Neuroscience:Official J Society for Neuroscience,2002,22(23):10494-10500.
[36] Bohn L M,Gainetdinov R R,Caron M G. G proteincoupled receptor kinase/beta-arrestin systems and drugs of abuse:psychostimulant and opiate studies in knockout mice[J]. Neuromolecular Medicine,2004,5(1):41-50.
[37] Kirsten M Raehal,Laura M Bohn. The role of betaarrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics[J]. Neuropharmacology,2011,60(1):58-65.
[38] Barhara Przewlocka,Agnieszka Sieja,Katarzyna Starowicz,et al. Knockdown of spinal opioid receptors by antisense targeting beta-arrestin reduces morphine tolerance and allodynia in rat[J]. Neuroscience Letters,2002,325(2):107-110.
[39] Cullen Schmid,Nicole M Kennedy,Nicolette C Ross,et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics[J]. Cell,2017,171(5):1165-1175.e1113.
[40] Zhou X Edward,He Yuan-zheng,Parker W de Waal,et al. Identification of phosphorylation codes for arrestin recruitment by g protein-coupled receptors[J]. Cell,2017,170(3):457-469.e413.
[41] Scott M DeWire,Dennis S Yamashita,David H Rominger,et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine[J]. J Pharmacology Experimental Therapeutics,2013,344(3):708-717.
[42] Neil Singla,Harold S Minkowitz,David G Soergel,et al.A randomized,Phase IIb study investigating oliceridine(TRV130),a novel micro-receptor G-protein pathway selective (mu-GPS) modulator,for the management of moderate to severe acute pain following abdominoplasty[J]. J Pain Research,2017,10:2413-2424.
[43] Aashish Manglik,Lin Henry,Dipendra Aryal,et al.Structure-based discovery of opioid analgesics with reduced side effects[J]. Nature,2016,537(7619):185-190.
[44] Neil Burford,Traynor J R,Alt A. Positive allosteric modulators of the mu-opioid receptor:a novel approach for future pain medications[J]. British J Pharmacology,2015,172(2):277-286.
[45] Martin C Michel,Steven J Charlton. Biased agonism in drug discovery-is it too soon to choose a path? [J].Molecular Pharmacology,2018,93(4):mol.117.110890.
